Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Chunzhang Yang and Version 2 by Vivi Li.
Mutations in isocitrate dehydrogenase (IDH) are commonly observed in lower-grade glioma and secondary glioblastomas. IDH mutants confer a neomorphic enzyme activity that converts α-ketoglutarate to an oncometabolite D-2-hydroxyglutarate, which impacts cellular epigenetics and metabolism. IDH mutation establishes distinctive patterns in metabolism, cancer biology, and the therapeutic sensitivity of glioma. Thus, a deeper understanding of the roles of IDH mutations is of great value to improve the therapeutic efficacy of glioma and other malignancies that share similar genetic characteristics.
- IDH mutation
- glioma
- cancer
- therapy resistance
1. Introduction
In 2008, compelling research showed that mutations in isocitrate dehydrogenase (IDH1 and IDH2) are frequently identified in the World Health Organization (WHO) grade II/III gliomas and secondary glioblastomas (GBMs). In contrast, these mutations are rare in primary GBM patients [1]. In 2009, Yan et al. [2] showed that IDH1 and IDH2 mutations frequently occur in WHO grade II/III astrocytomas and oligodendrogliomas. Besides gliomas, IDH mutations also occur in other non-central nervous system (CNS) malignancies, including acute myeloid leukemia (AML) [3][4][3,4], intrahepatic cholangiocarcinoma [5][6][5,6], chondrosarcoma [7], and melanoma [8][9][8,9]. The mutations are confined to a single arginine residue (Arg132) in IDH1 or two arginine residues (Arg172 and Arg140) in IDH2 [10][11][10,11]. The mutations commonly cause amino acid substitutions, which localize at the active sites of the enzymes and alter the catalytic functions of IDH enzymes. In contrast to wild-type IDH, which transforms isocitrate into α-ketoglutarate (α-KG), the mutated IDHs convert α-KG into D-2-hydroxyglutarate (D-2-HG) [12]. The altered catalytic activity that occurs because of cancer-associated IDH mutations was later termed “neomorphic activity”. The overproduction of the oncometabolite D-2-HG leads to widespread physiological consequences, including profound effects on cellular metabolism [13][14][13,14], epigenetic shift [15][16][17][18][15,16,17,18], genomic instability [19][20][21][22][23][19,20,21,22,23], and redox homeostasis [24][25][26][27][28][29][24,25,26,27,28,29]. IDH mutations are considered founder events for oncogenesis, through which an ancestor glial cell commits to malignant transformation. On the other hand, the mutant IDH enzyme brings about substantial changes in cancer biology, thereby establishing novel therapeutic vulnerabilities that are not commonly identified in other neoplasms.
2. Clinical Indications Involving the Discovery of IDH-Mutated Glioma
2.1. Clinical Classification of Gliomas
The 2016 WHO classification of CNS tumors has suggested the use of integrated phenotypic and genotypic characterization, which provides an increased level of objectivity [30][75]. In particular, IDH mutations have become some of the most important parameters in the differential diagnosis of gliomas. For example, diffuse astrocytomas often harbor IDH mutations, followed by other mutations such as TP53 and ATRX. Oligodendrogliomas are characterized by IDH mutations along with 1p/19q co-deletion (potentially along with CIC and FUBP1 mutations). The IDH mutation status is also useful for the differential diagnosis of primary and secondary GBMs [30][31][32][75,76,77]. Moreover, as IDH mutations frequently induce genome-wide DNA and histone hypermethylation, the introduction of methylation profiling allows for further improving the accuracy of glioma classification. Recently, Jaunmuktane et al. [33][78] demonstrated a diagnostic algorithm that integrated histology, molecular signature, and methylation array, and improved the diagnostic approach. Thus, the IDH mutation status is of great value in glioma classification and the selection of appropriate therapeutic strategies.
2.2. Radiology—D-2-HG Imaging
D-2-HG is a novel metabolite that accumulates in extremely high levels in glioma cells, but is absent in normal brain cells. The drastic contrast in cellular D-2-HG levels suggests that this oncometabolite could be an ideal biomarker for clinical monitoring and diagnosis among patients with IDH-mutated cancers [34][79]. Several hallmark studies have developed noninvasive radiologic methods for the detection of D-2-HG, such as magnetic resonance spectroscopy (MRS). In IDH mutant gliomas, D-2-HG accumulates to sufficient levels as a brain metabolite, which renders its visibility on MRS. These levels are 2–3 orders of magnitude higher than those found in the adjacent normal brain tissues [34][79]. Andronesi et al. [35][80] reported that D-2-HG was detected unambiguously in mutant IDH1 glioma in vivo using 2D correlation spectroscopy (COSY) and J-difference spectroscopy. Several other studies have also reported that D-2-HG is detected among glioma patients or in animal models using the short echo times (TEs) method [36][37][38][39][81,82,83,84]. On the other hand, D-2-HG levels were detected in glioma patients using long TE methods and J-difference spectroscopy with 100% sensitivity [40][41][85,86]. The application of long TE methods in D-2-HG detection has been confirmed in several subsequent reports, with increased sensitivity and specificity [42][43][87,88]. Overall, the noninvasive detection of D-2-HG has been proven to be a valuable diagnostic and prognostic biomarker. D-2-HG imaging provides a useful approach to the clinical management of patients with IDH-mutated glioma. Fluctuations in D-2-HG levels may provide crucial longitudinal data for the determination of disease progression and therapy response [34][79].
2.3. Disease Outcomes—Prolonged Survival
In 2008, Parsons et al. [1] first reported that mutations in IDH1 occurred in most patients with secondary GBM, and were associated with better overall survival (OS). Similar trends were reported in numerous studies using various datasets [44][45][46][47][48][49][42,89,90,91,92,93]. For example, using a large clinical dataset, Yan et al. [2] reported that GBM patients harboring IDH1 or IDH2 mutations tend to have a prolonged median OS compared with patients with IDH wild-type GBM. Similar findings were also observed among patients with anaplastic astrocytoma. The median OS was 65 months for patients with IDH mutant disease, compared with 20 months for those with IDH wild-type disease. Moreover, the progression-free survival (PFS) was also improved among GBM patients with IDH mutations compared with their counterparts [45][89]. Secondary genetic alterations, such as TP53/ATRX mutations and 1p/19q co-deletion, predispose patients with IDH-mutated gliomas to slightly different OS and disease-free survival (DFS; Figure 13A,B). Several studies have reported that IDH mutations are associated with younger age at diagnosis and limited genome alterations among patients with WHO grade II/III gliomas and GBMs, which may bias the disease outcome (Figure 13C,D) [1][2][50][51][1,2,94,95]. However, in a multivariate analysis, Sanson et al. [45][89] showed that the IDH mutation status is an independent predictor of favorable outcomes among glioma patients.
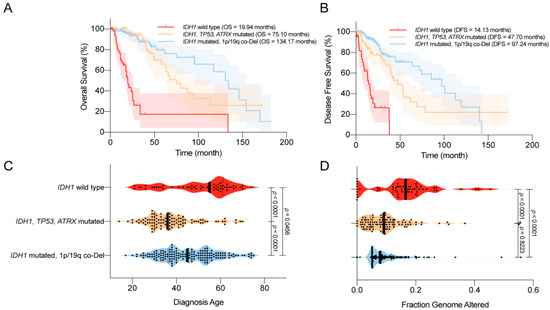
Figure 13. Clinical features of the World Health Organization (WHO) grade II/III IDH-mutated glioma. (A) Overall survival (OS) of glioma patients according to IDH1 status. IDH1 mutations are associated with prolonged OS. (B) Disease-free survival (DFS) of glioma patients according to IDH1 status. IDH1 mutations are associated with prolonged DFS. (C) Age at diagnosis among glioma patients according to IDH1 status. IDH1 mutations are associated with a younger age at diagnosis. (D) The distribution of genome alterations in glioma according to IDH1 status. IDH1 mutations are associated with fewer genome alterations. The data are visualized in cBioPortal [52][53][46,47].
2.4. Complications—Epilepsy and Secondary GBM
Epileptic seizure is one of the most common complications among patients with glioma, particularly those with LGGs (up to 90%) [54][55][56][57][96,97,98,99]. Severe seizures impair the quality of life and neurocognition function among glioma patients [58][100]. Considering the high incidence of IDH mutations in LGG, it is likely that the epileptic changes are relevant to the unique patterns in the tumor microenvironment, which is associated with IDH mutants. Numerous studies have indicated that mutations in IDH are associated with a high prevalence of epilepsy [59][60][61][62][63][101,102,103,104,105]. For example, Chen et al. [62][104] showed that IDH mutations are independently correlated with seizures, regardless of WHO grade. A recent study suggested that D-2-HG overproduction in the tumor microenvironment plays a major role in glioma-related epilepsy. D-2-HG is structurally similar to glutamate, which is the predominant excitatory neurotransmitter in the CNS. Thus, D-2-HG may act as an analog of glutamate, which leads to the abnormal firing of neurons through activating N-Methyl-d-aspartic acid (NMDA) receptors, and hence epileptic changes. Treating cultured rat cortical neurons with exogenous D-2-HG resulted in an elevated firing rate [62][104]. By mimicking the activity of glutamate, the increased level of D-2-HG mediates the abnormal neuronal activity and leads to glioma-related epilepsy [35][64][65][80,106,107]. However, three millimolar D-2-HG induced an elevated burst frequency in the neuronal network in vitro [62][104], whereas this dose is over 30 times higher than the glutamate concentration for excitotoxicity [66][108]. More effort is urged in order to elucidate the detailed molecular mechanism of the epileptic changes in IDH-mutated glioma. Because of the association between IDH mutations and seizures, therapies that target mutant IDH, such as mutant IDH inhibitors, could diminish D-2-HG production and potentially reduce epileptic seizures [67][109].
2.5. Sensitivity to Radiotherapy and Chemotherapy
Clinical data have shown that IDH mutant gliomas tend to exhibit a better disease outcome compared with wild-type IDH tumors. Several studies have explained that the favorable prognosis of IDH mutant gliomas is due to their increased sensitivity to radiotherapy and chemotherapy [68][69][110,111]. IDH mutant gliomas likely harbor defects in multiple DNA repair pathways, which render them vulnerable to radiotherapy- or chemotherapy-induced DNA damage [19][22][19,22]. These findings indicate that IDH mutation could serve as an important predictive factor for treatment response among glioma patients. For example, Houillier et al. [69][111] reported that IDH1 mutation is an independent predictor of temozolomide response among LGG patients. IDH1 mutations combined with 1p/19q co-deletion further improved the treatment response. Hartman et al. [70][112] also reported that IDH1 status is an important predictor of disease-free survival (DFS) and OS among patients undergoing adjuvant therapy. In another study conducted by van de Bent et al. [71][113], no correlation was found between IDH1 mutations and disease outcome in response to procarbazine (Matulane), lomustine (CCNU), and vincristine (Oncovin) chemotherapy.
3. Novel Molecular Targeting for IDH-Mutated Glioma
3.1. IDH Mutant Inhibitors
Because of the critical roles played by IDH mutations in the malignant transformation of glioma, targeting the neomorphic activity of IDH mutants has been heavily proposed as a direct therapeutic approach. In the past decade, several attempts have been made to develop small molecular compounds that directly inhibit mutant IDH enzymes. In 2012, the first-in-class mutant IDH inhibitor was discovered, which showed a specific and potent inhibitory effect on D-2-HG production in IDH mutant U87 cells and xenograft models [72][114]. Later, Rohle et al. [67][109] reported a novel synthetic inhibitor of IDH mutant, AGI-5198, which blocked D-2-HG production and subsequently reversed the malignant transformation effect of IDH mutations. Besides glioma, the inhibition of mutant IDH promotes differentiation in leukemia harboring IDH mutations [6]. With the promising findings regarding AGI-5198, second-generation mutant IDH inhibitors are under development and are undergoing evaluation in clinical studies. For example, ivosidenib (AG-120) and vorasidenib (AG-881) have been tested in AML and glioma with IDH mutations [73][74][75][76][115,116,117,118]. In a recent phase I clinical study with ivosidenib in IDH1-mutated advanced glioma conducted by Mellinghoff et al. [77][119], the mutant IDH inhibitor appeared to be well-tolerated throughout the experiment, which paved the way for subsequent clinical studies to evaluate its therapeutic efficacy. Although the IDH mutant enzyme inhibitors suppress malignancy, several studies have suggested that this inhibitor reduces D-2-HG production and relieves the burden on the DNA repair pathway, resulting in chemoresistance to other therapies, such as PARP inhibitors [23][78][23,120]. More effort is urged to explore the strategy of combining IDH mutant inhibitors with other glioma therapies in order to improve the clinical outcome.
3.2. Targeting Hypermethylation Phenotype
Genome-wide DNA and histone hypermethylation is a unique signature in IDH-mutated glioma, which is closely related to gliomagenesis by promoting oncogene expression and inhibiting tumor suppressors [79][63]. This rectification of the epigenetic shift could be a reasonable strategy for halting D-2-HG-driven oncogenesis and the malignant phenotype. DNA-demethylating agents such as 5-azacytidine or 5-aza-2′-deoxycytidine (decitabine) irreversibly bind to DNA methyltransferases (DNMTs) and inhibit the process of DNA methylation. The D-2-HG-induced hypermethylation phenotype was reversed by demethylating compounds, and cell proliferation was suppressed in vitro and in vivo [80][81][82][121,122,123]. Several clinical trials are evaluating the therapeutic effects of 5-azacytidine among patients with recurrent gliomas with IDH mutations (NCT03666559 and NCT03684811). On the other hand, inhibitors targeting histone methyltransferases inhibitors are also being investigated for IDH-mutated gliomas, as an alternative strategy to rectify the D-2-HG-associated hypermethylation phenotype. It is reported that H3K9 methyltransferase G9a is correlated to the development and progression of glioma, and its inhibitor BIX-01294 showed repressive effects on gliomas cells [83][124].
3.3. Targeting DNA Repair Pathways
As previously mentioned, IDH mutant gliomas exhibit defects in multiple DNA repair pathways. High levels of D-2-HG inhibit the activity of DNA oxidative demethylases, such as AlκB homolog 2/3 (ALKBH2/3) [19]. Several seminal studies have also indicated that D-2-HG compromises HR DNA repair, establishing a “BRCAness” in this type of malignancy [23][84][23,125]. In addition, IDH mutation-associated G-CIMP resulted in the methylation of the promoter region of O-6-methylguanine-DNA methyltransferase (MGMT), which reduced MGMT expression and led to increased sensitivity to alkylating agents [46][85][86][90,126,127]. Our recent study indicated that IDH mutations led to defects in NAD metabolism, which compromised PARP-associated HR, as PARP repairs DNA damage in an NAD+ dependent manner [22][87][22,69]. With the identification of the DNA repair deficiency in IDH-mutated glioma, numerous studies have attempted to evaluate DNA repair inhibitors, which may serve as a potential sensitization strategy. Several other groups and as well as ours reported that a combination of PARP inhibitors, such as olaparib, with temozolomide or radiotherapy, led to synergistic lethality in IDH mutant glioma cells [21][22][23][21,22,23]. Several phase I/II clinical trials are currently recruiting patients to investigate the therapeutic effect of the PARP inhibitors, pamiparib (BGB-290) or olaparib, combined with temozolomide in IDH mutant gliomas (NCT03914742, NCT03749187, and NCT03212274).
3.4. Targeting Anti-Oxidative Pathways
Redox homeostasis has been reported to be greatly impacted by IDH mutations, highlighted by profoundly elevated levels of oxidative stress [24][25][26][27][28][24,25,26,27,28]. As a result, ROS scavenging pathways are widely mobilized in the context of IDH mutation, so as to maintain cellular metabolism, thereby supporting cellular growth and survival. These findings suggest that the antioxidant pathway plays an essential role in IDH-mutated glioma. Targeting anti-oxidative pathways may be more effective in glioma with IDH mutations. Our recent study showed that NRF2-governed anti-oxidative pathways, such as that regarding de novo glutathione synthesis, were widespread in IDH mutant gliomas. The blockade of NRF2 using natural compound inhibitors, brusatol, or triptolide significantly increased oxidative damage and subsequently suppressed the growth of IDH mutant xenografts with prolonged OS [25][27][28][88][25,27,28,128]. The concept of targeting redox homeostasis in IDH mutant cancers has shown a potential therapeutic value. The development of pharmacological grade NRF2 inhibitors is needed urgently for potential clinical translation.
3.5. Targeting Metabolic Reprogramming
D-2-HG is a metabolite that is absent in normal cells. The production of large quantities of D-2-HG inevitably depletes a substantial amount of carbohydrate from the Krebs cycle. Several hallmark studies have demonstrated the presence of depleted metabolic pathways in IDH-mutated cells. For example, glutamate metabolism is greatly altered in IDH mutant glioma, as mentioned before. The glutamate level is significantly lower in IDH mutant cancers, which leads to an increased dependence on glutaminolysis to compensate for the metabolism [29][89][90][91][29,74,129,130]. Several studies have reported that a blockade of glutaminase activity results in the suppression of IDH mutant glioma and AML. Seltzer et al. and Emadi et al. [89][90][74,129] reported that bis-2-(5-phenylacetamido-1,2,4-thiadiazol-2-yl) ethyl sulfide (BPTES), an inhibitor of glutaminase, selectively suppresses tumor growth in IDH mutant glioma and AML by targeting the fragile glutamine metabolism. Another glutaminase inhibitor (CB-839) was also reported to induce selective radio-sensitivity in IDH mutant cancers [29] and terminal differentiation in IDH mutant AML [91][130]. An ongoing phase I clinical trial is investigating the side effects and the best dose of CB-839, in combination with radiation therapy and temozolomide, for treating IDH-mutated diffuse or anaplastic astrocytoma (NCT03528642). In addition, IDH mutations lead to the depletion of NAD+ because of the increased methylation of the promoter region of NAPRT1, the rate-limiting enzyme in NAD+ biosynthesis, and suppression of the expression of NAPRT1. This renders the IDH mutant glioma vulnerable to inhibition through the nicotinamide phosphoribosyltransferase (NAMPT) catalyzed NAD+ salvage pathway [92][131]. Moreover, Tateishi et al. [93][132] showed that NAMPT inhibitors further sensitized IDH mutant cancer cells to alkylating agents, such as temozolomide, as PARP activation consumes NAD+ during the base excision repair of chemotherapy-induced DNA damage. With the substantially exhausted metabolic pathways, distinctive metabolic vulnerabilities are established in IDH-mutated malignancies. Effectively targeting these metabolic pathways may induce selective cytotoxicity to cancer cells, but a lesser extent than that occurs in normal somatic cells with an intact metabolic network.
Acknowledgment
This work was supported by the Intramural Research Program of the National Institutes of Health,
National Cancer Institute.
National Cancer Institute.