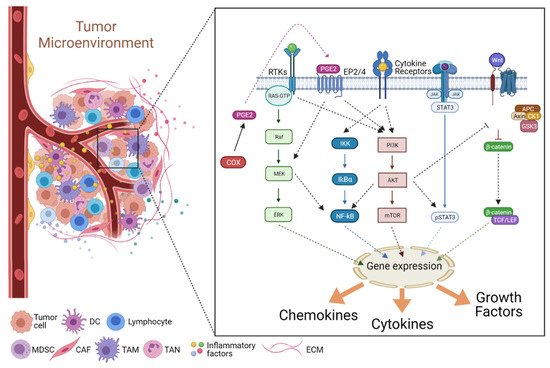
The development of tumors requires an initiator event, usually exposure to DNA damaging agents that cause genetic alterations such as gene mutations or chromosomal abnormalities, leading to deregulated cell proliferation. Although the mere stochastic accumulation of further mutations may cause tumor progression, it is now clear that an inflammatory microenvironment has a major tumor-promoting influence on initiated cells, in particular when a chronic inflammatory reaction already existed before the initiated tumor cell was formed. Moreover, inflammatory cells become mobilized in response to signals emanating from tumor cells. In both cases, the microenvironment provides signals that initiated tumor cells perceive by membrane receptors and transduce via downstream kinase cascades to modulate multiple cellular processes and respond with changes in cell gene expression, metabolism, and morphology. Cytokines, chemokines, and growth factors are examples of major signals secreted by immune cells, fibroblast, and endothelial cells and mediate an intricate cell-cell crosstalk in an inflammatory microenvironment, which contributes to increased cancer cell survival, phenotypic plasticity and adaptation to surrounding tissue conditions. Eventually, consequent changes in extracellular matrix stiffness and architecture, coupled with additional genetic alterations, further fortify the malignant progression of tumor cells, priming them for invasion and metastasis.
- tumor microenvironment
- inflammation
- signal transduction
- cancer
Note: The following contents are extract from your paper. The entry will be online only after author check and submit it.
1. Introduction
2. Cancer-Associated Inflammation (CAI) and the TME
2.1. Mediators of Cancer-Associated Inflammation (CAI)
2.1.1. Pro-Inflammatory Factors
2.1.2. Pro-Inflammatory Chemokines in the TME
2.1.3. Anti-Inflammatory Mediators—IL-10
2.1.4. The Transforming Growth Factor-β (TGF-β)
2.2. Signaling in the Inflammatory TME