Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Rita Xu and Version 1 by CHUNYI WEN.
As a hallmark of osteoarthritis (OA), structural and functional disturbances of the subchondral bone are associated with osteocyte dysfunction. Osteocytes act as mechanosensory units for micro-cracks in response to loading, and as the regulator of osteoblastic and osteoclastic activities. Aberrant expressions of osteocyte-derived signaling molecules likely underlie the anomalies seen in osteocyte morphology and bone remodelling.
- osteoarthritis
- osteocyte
- bone remodeling
- articular cartilage
- sclerostin
1. Introduction
Osteoarthritis (OA) is the most common form of arthritic disease, mainly afflicting the load-bearing joints, e.g., knee and hip. It is also recognized as a major cause of joint pain and disability, contributing to a compromised quality of life in older adults [1,2][1][2]. Articular cartilage, subchondral bone, and synovium are among the tissues that may display abnormalities in OA. The radiological features of osteoarthritic subchondral bone include sclerosis, cyst and osteophyte formation on plain x-radiograph, and bone marrow lesions (BMLs) on magnetic resonance imaging, all of which have been proven to underline the anomalies of bone mineralization [3,4][3][4]. Such impaired mineralization was noted from all parts of the trabecular bone, along with compositional changes of the bone matrix, e.g., a low ratio of mineral/collagen content [5,6][5][6]. Sclerotic changes and osteophyte formation are both believed to arise from elevated bone turnover with an increase in osteoblastic over osteoclastic activities [7,8][7][8]. BMLs and sclerosis are likely associated with abnormal signaling related to sclerostin, periostin, and dentin matrix protein 1 (DMP-1) [9,10,11][9][10][11]. Meanwhile, cysts surrounded by less mineralized bone and osteoid formation uncoupled with mineralization may suggest spatially differential regulation of osteoblasts and osteoclasts by Wnt/β-catenin signaling and the OPG/RANKL/RANK pathway [12,13][12][13]. Modulation of bone remodeling was able to reduce osteophyte formation and alleviate cartilage degeneration in experimental models of OA and helped with restricting BMLs in human OA [14,15,16][14][15][16]. However, the pathomechanism of osteoarthritic bone remodeling and the resultant structural disturbances has not been fully understood yet.
Bone resorption and formation are tightly controlled and precisely coordinated by bone cells during the bone remodeling process to maintain the homeostasis of adult bone metabolism [17]. The structural and functional disturbances of the osteoarthritic bone are generally attributed to primary osteoblastic dysfunction [18]. Osteoarthritic osteoblasts showed blunted responses to the stimulus of cytokines [19,20][19][20] and an aberrant ratio of mineral and collagen production [6]. Meanwhile, osteocyte dysfunction and abnormal osteoblast-osteocyte differentiation are also recognized as the possible causes of OA [21]. Osteocytes compose 90–95% of bone cells and are responsible for mechano-transduction. It is known that osteocytes may respond to mechanical stimuli like micro-cracks with death. Following the deaths of osteocytes, bone remodeling is initiated via activation of osteoclastogenesis and bone resorption [17]. Osteocytes also act as the key regulators of osteoblast differentiation through the secretion of sclerostin. Through suppressing Wnt signaling, sclerostin can inhibit osteoblastic activity and terminate the cycle of bone remodeling [17,22,23][17][22][23]. There is growing evidence suggesting that osteocytes act as a central regulator of bone remodeling to maintain bone homeostasis and integrity [24]. However, the exact pathophysiology of osteocytes and their contributions to the structural and functional disturbances of the osteoarthritic bone remain unelucidated.
2. Disturbances of Subchondral Bone
The role of subchondral bone in OA progression has been well documented. Disturbances of the subchondral bone are characterized by anomalous bone morphology at the tissue level, irregular osteocyte activities at the cellular level, and altered protein expressions at the molecular level. The Wnt/β-catenin pathway, which is responsible for osteogenesis and bone remodeling, is enriched in osteoarthritic subchondral bones [12]. Sclerostin, a major Wnt inhibitor, is expressed in calcified cartilage and subchondral bone. Its deficiency is often associated with OA development, likely through activating Wnt signaling [25]. In both OA and sclerostin-deficient models, an increase in bone volume fraction and trabecular bone thickness were observed in the subchondral bone, along with aberrant distribution of bone mineral density (BMD) [25,26,27,28][25][26][27][28]. This change is believed to be load-dependent, as higher bone mass is usually found excessively concentrated in the load-bearing region, with relatively lower bone mass in the less or non-loading region. For instance, our data suggested that osteoarthritic bone exhibited unevenly distributed bone mass from the load-bearing to non-load-bearing regions, such as in the superior portion of femoral heads underneath the articular cartilage. In contrast, there was no significant difference in the bone mass organization of the femoral heads from the control group (Figure 1a,b). In accordance with structural changes, sclerostin levels are also believed to be modulated by loading. Expression of sclerostin by osteocytes was downregulated by mechanical loading while upregulated by unloading in mice, suggesting a role in mechano-transduction [29,30][29][30]. Overexpression of SOST, the gene encoding sclerostin, was found to inactivate Wnt signaling, attenuate bone formation, and reduce mineralization in response to mechanical stimulation (Figure 1c). Suppressed bone remodeling then resulted in little to no difference in load-bearing and non-loading regions [30]. Hence, changes in sclerostin levels likely underlie the occurrence of sclerotic lesions preferentially in load-bearing regions. It might thus be considered as an adaption to support an abnormal level of compressive stress, such as when cartilage tissues are severely eroded [26,31][26][31] or when repetitive loading is applied [32].
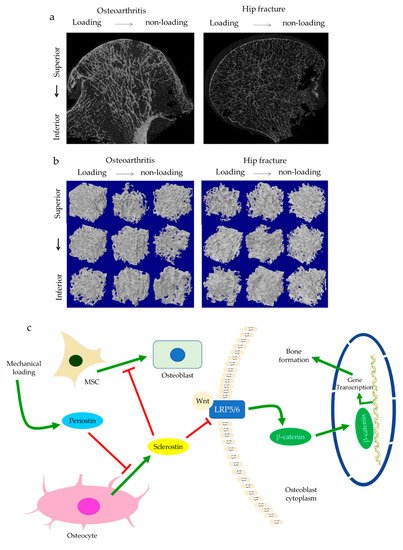
Figure 1. The representative uCT images and data show the comparisons of hip osteoarthritis (OA) with a fracture in two- (a) and three-dimensional architecture (b). Osteoarthritic bone was characterized by high bone mass disproportionately concentrated in the load-bearing regions in comparison with hip fracture bone. This is believed to be regulated by periostin and sclerostin (c). Enhanced periostin and decreased sclerostin expressions upon mechanical loading can activate Wnt/β-catenin signaling through LRP5/6. β-catenin then promotes gene transcription in the targeted cell, such as osteoblasts, which results in bone formation.
Subchondral trabeculae are generally well-aligned with the direction of principal compressive stress, suggesting adaptation to loading conditions [26,27][26][27]. Periostin, a matricellular protein produced by osteocytes and osteoblasts, may participate in the regulation of bone formation and resorption associated with sclerostin. Increased periostin expression was observed upon mechanical stimulation, which was followed by a decreased sclerostin expression (Figure 1c) [10,33][10][33]. Periostin expression was enhanced in OA in subchondral bone, synovial fluid, and cartilage [34,35,36][34][35][36]. It was thought to exacerbate OA by promoting the expressions of pro-inflammatory cytokines, such as interleukin (IL)-6 and -8, and matrix metalloproteinase (MMP) production, such as through nuclear factor kappa B (NFκB) signaling [35]. High periostin level was linked with decreased bone surface/bone volume ratio, increased trabecular number, low structural model index (SMI), and high stiffness [10,11][10][11]. Such changes echoed the findings in OA, specifically the drop in bone surface area, the elevated number of trabeculae bones and their plate-like microarchitecture, as well as the stiffening of subchondral bones [26,27][26][27]. Plate-like trabecular bones were positively correlated with OA severity [37] and were generally able to sustain higher mechanical stress [38]. Meanwhile, stiffening of subchondral bone could cause ineffective load transfer from cartilage to bone. This was proposed as a possible source of cartilage damage and might explain the exacerbation of OA under excessive loading [39]. Thus, periostin might also induce cartilage degeneration by disturbing the underlying subchondral bone through sclerostin-Wnt signaling [11,40][11][40]. Interestingly, other characteristics of osteoarthritic bones, such as decreased trabeculae connectivity and BMD, were identified in periostin-deficient models [10,11][10][11]. Likewise, inhibition of sclerosing activities through antibodies resulted in higher connectivity density and BMD, which appeared to contradict the findings in OA [41,42][41][42]. Nevertheless, it should be noted that hyper-mineralization of the subchondral bone was also observed in early-stage OA [43]. Therefore, periostin and sclerostin might first affect bone mineralization in early-stage OA, while impaired mineralization in end-stage OA could relate to cellular dysfunction. Other signaling pathways might also be responsible for these sclerotic changes [44], such as transforming growth factor-beta (TGF-β) signaling [45], whose inhibition increased connectivity density and attenuated OA, DMP-1, whose deletion in cartilage led to an osteoarthritic phenotype [9,46][9][46].
Subchondral bone marrow lesions (BMLs) have been characterized by sclerotic bones with less mineralization [3] and are believed to predict the progression of OA [47,48,49,50][47][48][49][50]. Our data also confirmed that the inorganic content of bone plugs from knee OA specimens was significantly lower (Figure 2a,b). We have also observed osteoid formation near trabeculae or in the marrow space of OA bone (Figure 3g,i), which were compatible with that of BMLs. Aberrant mineralization is likely associated with DMP-1. DMP-1 is responsible for the mineralization of collagen content, as it is critical for mineral nucleation [51]. Decreased DMP-1 expression and disrupted mineralization of the extracellular matrix (ECM) is accompanied by the activation of Wnt/β-catenin signaling, along with sclerostin deficiency [52]. Altered DMP-1 signaling might explain why OA bone exhibited higher mass yet with decreased structural connectivity and reduced mineral density. However, DMP-1 expression by osteocytes was higher in human OA samples [28]. Meanwhile, mechanical loading appeared to upregulate DMP-1 expression by osteocytes in both bone formation and resorption sites, while osteoblasts expressed less DMP-1 in the early stage of induction [53]. Hence, decreased DMP-1 expression in osteoblasts was proposed as a possible cause of low BMD in OA. This was supported by the results of our research, which showed the formation of osteoid-like tissues in marrow space together with the clustering of bone cells in OA (Figure 3i). Low BMD and poor connectivity were associated with decreased strength. Despite being plate-like and well-aligned, osteoarthritic bones were also weaker than normal bones with similar morphology [54,55][54][55]. Therefore, although the initial attempt was to increase BMD in response to mechanical loading, bone remodeling in OA might eventually render the subchondral bone mechanically inferior rather than superior.
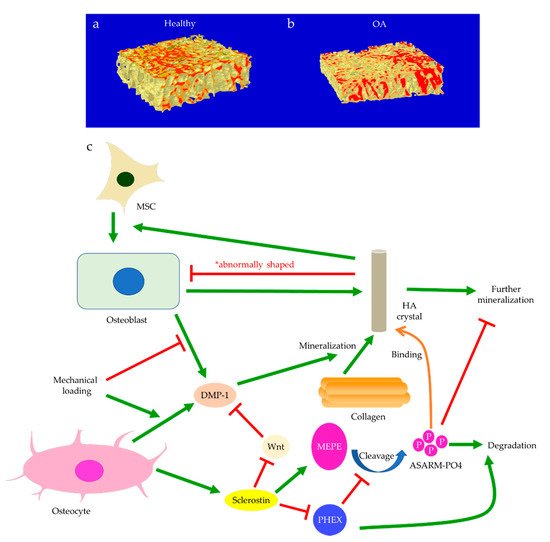
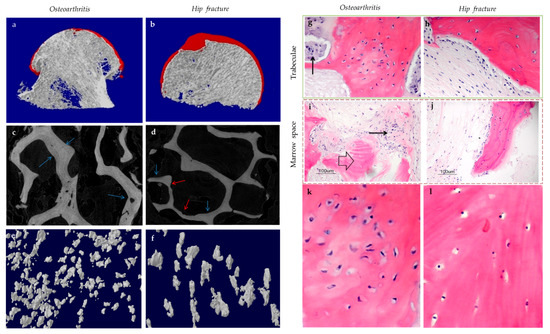
Figure 2. The representative micro-CT images show the microstructure of trabecular bone in the medial plateau of healthy (a) and osteoarthritic (OA) knees (b). The color-coded images of trabecular bone show the distribution of bone mineral density (BMD). The red-labeled trabecular bone represented high BMD. Bone mineralization is regulated by sclerostin, dentin matrix protein 1 (DMP-1), matrix extracellular phosphoglycoprotein (MEPE), and phosphate-regulating neutral endopeptidase (PHEX) (c). Osteoblast-derived DMP-1 is critical for the mineralization of collagen fibers, while sclerostin can regulate MEPE and PHEX to control further mineralization of hydroxyapatite (HA) crystals. Mechanical loading regulates DMP-1 expressions by osteoblasts and osteocytes differently. Abnormally shaped HA crystals can induce osteogenic differentiation of mesenchymal stem cells (MSCs) or trigger osteoblast dysfunction.
Figure 3. The representative uCT (a–f) and histological images (g–l) show the comparisons between hip osteoarthritis (OA) (a,c,e,g,i,k) and fracture (b,d,f,h,j,l). As shown by uCT images, osteoid formation (blue arrow) was found in apposition to pre-existing trabeculae for both hip OA (c) and fracture (d). The newly formed bone was associated spatially with bone resorption pits (red arrow) in hip fracture, yet it was not the case for hip OA. It was revealed by histology that bone cell clusters (black arrow) were present close to osteoid formation (block arrow) either in trabecular bone (g) or marrow space (i). In contrast, bone cell clusters were not found in hip fractures (h,j). The two (k,l) and three (e,f) dimensional morphology of osteocytes lacunae both revealed that the number of lacunae from hip OA bone was higher than that of hip fracture. In addition, OA osteocytes lacunae were well aligned and more plate-like as compared with hip fracture ones. (g–j: Hematoxylin and eosin staining with a magnification of 20×).
Crosstalk with Cartilage Degeneration
Disturbances of the subchondral bone have been closely associated with cartilage degeneration. For example, the size of the tibial subchondral bone might predict the overlying articular cartilage defect and OA severity [56]. Insulin-like growth factor 1 (Igf1) deficiency was linked with decreased bone formation, increased osteoid formation, and delayed and diminished mineral deposition in the subchondral bone (Figure 4) [57]. In OA, Igf1 expression by osteoblasts was upregulated [19,36][19][36]. Similarly, the mechanical strain was believed to increase Igf1 levels in osteocytes [58,59][58][59]. Hence, Igf1 was thought to facilitate the drop in sclerostin level upon mechanical loading and thus modulate bone remodeling [60]. In contrast, Igf1 expression by osteoarthritic chondrocytes was reduced [61]. This might be attributed to the presence of stressors, such as reactive oxygen species (ROS) [62], a well-identified cause of cartilage damage [63]. The addition of Igf1 to articular cartilage could increase matrix biosynthesis or enhance the stimulating effects of dynamic mechanical loading [64]. Likewise, matrix degradation in response to static loading could also be rescued by Igf1, which might suggest a role of Igf1 in maintaining the balance of anabolism and catabolism [65]. Cartilage degeneration is thus likely linked with both decreased anabolic activities partly due to Igf1 deficiency and increased catabolism associated with elevated expressions of degrative enzymes, such as MMPs [28].
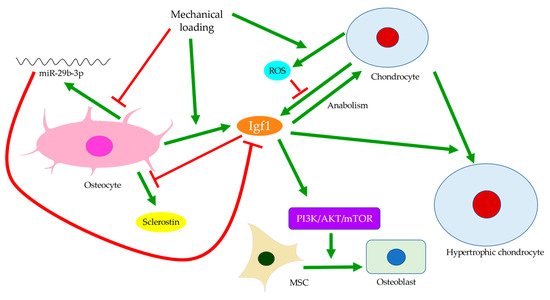
Figure 4. Insulin-like growth factor 1 (Igf1) signaling in the crosstalk between osteocytes and chondrocytes. Mechanical loading regulates Igf1 expressions by osteocytes and chondrocytes differently. Inhibition of miR-29b-3p production by osteocytes can increase Igf1 expression. Meanwhile, elevated reactive oxygen species (ROS) production by chondrocytes can suppress Igf1 expression. Igf1 also regulates sclerostin expression, chondrocyte hypertrophy, and osteogenic differentiation of MSCs through the PI3K/AKT/mTOR pathway.
Bone remodeling mediated by Wnt signaling is believed to stiffen trabecular bones and alter their micro-architecture. For instance, in spontaneous OA models of guinea pigs, changes from rod-like to plate-like morphology were thought to precede or occur simultaneously with cartilage degeneration [66,67][66][67]. Past experiments also observed subchondral bone remodeling beneath intact cartilage in human OA, but only significant when cartilage tissues were damaged [26,66][26][66]. Ineffective load transfer due to structural disturbances of the subchondral bone might increase the strain endured by cartilage. High mechanical strain could then increase ROS production by chondrocytes, presumably because of mitochondrial deformation (Figure 4) [68,69][68][69]. Therefore, differential expressions of Igf1 in bone and cartilage, for instance, could be involved in the progression of OA. Similarly, while TGF-β could promote chondrocyte proliferation and exhibited protective effects in articular cartilage, its elevation in the subchondral bone was associated with cartilage loss and OA development [45]. The crosstalk between subchondral bone remodeling and cartilage degeneration was also believed to be regulated by MMP-13, which was less expressed by osteoarthritic osteocytes. Insufficient expression of MMP-13 was assumed to increase trabecular bone volume and cause cartilage degeneration independent of other stimuli. A reduction in osteocyte-derived MMP-13 was associated with disrupted lacuno-canalicular networks (LCN), which likely affected mechano-transduction and bone remodeling in early-stage OA. Meanwhile, expressions of proteins responsible for metabolism or matrix degradation, such as MMP-13 expression by chondrocytes, were also induced by insufficient osteocytic MMP-13 [43]. Hence, bone remodeling might start before cartilage degeneration and contribute to the aberrant metabolism in cartilage. With accumulated micro-damage, such as when an excessive load was applied or when cartilage was lost [66], mechanically inferior bones would be formed predominantly, and OA exacerbated. The above findings suggested that osteocytes could regulate trabecular bone morphology, bone mineralization, and ultimately bone and cartilage homeostasis in response to mechanical loading. Hence, the interplay between cartilage and subchondral bone is worth further investigation and should be extended to cellular and protein levels.
References
- Kendzerska, T.; Juni, P.; King, L.K.; Croxford, R.; Stanaitis, I.; Hawker, G.A. The longitudinal relationship between hand, hip and knee osteoarthritis and cardiovascular events: A population-based cohort study. Osteoarthr. Cartil. 2017, 25, 1771–1780.
- Brandt, K.D.; Dieppe, P.; Radin, E. Etiopathogenesis of osteoarthritis. Med. Clin. N. Am. 2009, 93, 1–24.
- Hunter, D.J.; Gerstenfeld, L.; Bishop, G.; Davis, A.D.; Mason, Z.D.; Einhorn, T.A.; Maciewicz, R.A.; Newham, P.; Foster, M.; Jackson, S.; et al. Bone marrow lesions from osteoarthritis knees are characterized by sclerotic bone that is less well mineralized. Arthritis Res. Ther. 2009, 11, R11.
- Chiba, K.; Nango, N.; Kubota, S.; Okazaki, N.; Taguchi, K.; Osaki, M.; Ito, M. Relationship between microstructure and degree of mineralization in subchondral bone of osteoarthritis: A synchrotron radiation µCT study. J. Bone Miner. Res. 2012, 27, 1511–1517.
- Coats, A.M.; Zioupos, P.; Aspden, R.M. Material properties of subchondral bone from patients with osteoporosis or osteoarthritis by microindentation testing and electron probe microanalysis. Calcif. Tissue Int. 2003, 73, 66–71.
- Couchourel, D.; Aubry, I.; Delalandre, A.; Lavigne, M.; Martel-Pelletier, J.; Pelletier, J.-P.; Lajeunesse, D. Altered mineralization of human osteoarthritic osteoblasts is attributable to abnormal type I collagen production. Arthritis Rheumatol. 2009, 60, 1438–1450.
- Bettica, P.; Cline, G.; Hart, D.J.; Meyer, J.; Spector, T.D. Evidence for increased bone resorption in patients with progressive knee osteoarthritis: Longitudinal results from the Chingford study. Arthritis Rheumatol. 2002, 46, 3178–3184.
- Dai, G.; Xiao, H.; Liao, J.; Zhou, N.; Zhao, C.; Xu, W.; Xu, W.; Liang, X.; Huang, W. Osteocyte TGFβ1-Smad2/3 is positively associated with bone turnover parameters in subchondral bone of advanced osteoarthritis. Int. J. Mol. Med. 2020, 46, 167–178.
- Zhang, Q.; Lin, S.; Liu, Y.; Yuan, B.; Harris, S.E.; Feng, J.Q. Dmp1 null mice develop a unique osteoarthritis-like phenotype. Int. J. Biol. Sci. 2016, 12, 1203–1212.
- Bonnet, N.; Standley, K.N.; Bianchi, E.N.; Stadelmann, V.; Foti, M.; Conway, S.J.; Ferrari, S.L. The matricellular protein periostin is required for sost inhibition and the anabolic response to mechanical loading and physical activity. J. Biol. Chem. 2009, 284, 35939–35950.
- Lv, J.; Sun, X.; Ma, J.; Ma, X.; Xing, G.; Wang, Y.; Sun, L.; Wang, J.; Li, F.; Li, Y. Involvement of periostin-sclerostin-Wnt/β-catenin signaling pathway in the prevention of neurectomy-induced bone loss by naringin. Biochem. Biophys. Res. Commun. 2015, 468, 587–593.
- Funck-Brentano, T.; Bouaziz, W.; Marty, C.; Geoffroy, V.; Hay, E.; Cohen-Solal, M. Dkk-1-Mediated inhibition of Wnt signaling in bone ameliorates osteoarthritis in mice. Arthritis Rheumatol. 2014, 66, 3028–3039.
- Palumbo, C.; Ferretti, M. The osteocyte: From “prisoner” to “orchestrator”. J. Funct. Morphol. Kinesiol. 2021, 6, 28.
- Carbone, L.D.; Nevitt, M.C.; Wildy, K.; Barrow, K.D.; Harris, F.; Felson, D.; Peterfy, C.; Visser, M.; Harris, T.B.; Wang, B.W.E.; et al. The relationship of antiresorptive drug use to structural findings and symptoms of knee osteoarthritis. Arthritis Rheumatol. 2004, 50, 3516–3525.
- Ding, M.; Danielsen, C.C.; Hvid, I. The Effects of bone remodeling inhibition by alendronate on three-dimensional microarchitecture of subchondral bone tissues in guinea pig primary osteoarthrosis. Calcif. Tissue Int. 2008, 82, 77–86.
- Hayami, T.; Pickarski, M.; Wesolowski, G.A.; Mclane, J.; Bone, A.; Destefano, J.; Rodan, G.A.; Duong, L.T. The role of subchondral bone remodeling in osteoarthritis: Reduction of cartilage degeneration and prevention of osteophyte formation by alendronate in the rat anterior cruciate ligament transection model. Arthritis Rheumatol. 2004, 50, 1193–1206.
- Henriksen, K.; Neutzsky-Wulff, A.V.; Bonewald, L.F.; Karsdal, M.A. Local communication on and within bone controls bone remodeling. Bone 2009, 44, 1026–1033.
- Lajeunesse, D.; Reboul, P. Subchondral bone in osteoarthritis: A biologic link with articular cartilage leading to abnormal remodeling. Curr. Opin. Rheumatol. 2003, 15, 628–633.
- Massicotte, F.; Aubry, I.; Martel-Pelletier, J.; Pelletier, J.-P.; Fernandes, J.; Lajeunesse, D. Abnormal insulin-like growth factor 1 signaling in human osteoarthritic subchondral bone osteoblasts. Arthritis Res. Ther. 2006, 8, R177.
- Sanchez, C.; Deberg, M.A.; Bellahcène, A.; Castronovo, V.; Msika, P.; Delcour, J.P.; Crielaard, J.M.; Henrotin, Y.E. Phenotypic characterization of osteoblasts from the sclerotic zones of osteoarthritic subchondral bone. Arthritis Rheumatol. 2008, 58, 442–455.
- Hemmatian, H.; Bakker, A.D.; Klein-Nulend, J.; van Lenthe, G.H. Aging, osteocytes, and mechanotransduction. Curr. Osteoporos. Rep. 2017, 15, 401–411.
- Ten Dijke, P.; Krause, C.; de Gorter, D.J.J.; Löwik, C.W.G.M.; van Bezooijen, R.L. Osteocyte-Derived sclerostin inhibits bone formation: Its role in bone morphogenetic protein and Wnt signaling. J. Bone Jt. Surg. 2008, 90, 31–35.
- Van Bezooijen, R.L.; Roelen, B.A.J.; Visser, A.; van der Wee-Pals, L.; de Wilt, E.; Karperien, M.; Hamersma, H.; Papapoulos, S.E.; ten Dijke, P.; Löwik, C.W.G.M. Sclerostin is an osteocyte-expressed negative regulator of bone formation, but not a classical BMP antagonist. J. Exp. Med. 2004, 199, 805–814.
- Bonewald, L.F. Osteocytes: A proposed multifunctional bone cell. J. Musculoskelet. Neuronal Interact. 2002, 2, 239–241.
- Li, J.; Xue, J.; Jing, Y.; Wang, M.; Shu, R.; Xu, H.; Xue, C.; Feng, J.; Wang, P.; Bai, D. SOST deficiency aggravates osteoarthritis in mice by promoting sclerosis of subchondral bone. Biomed. Res. Int. 2019, 2019, 7623562.
- Chappard, C.; Peyrin, F.; Bonnassie, A.; Lemineur, G.; Brunet-Imbault, B.; Lespessailles, E.; Benhamou, C.L. Subchondral bone micro-architectural alterations in osteoarthritis: A synchrotron micro-computed tomography study. Osteoarthr. Cartil. 2006, 14, 215–223.
- Chiba, K.; Ito, M.; Osaki, M.; Uetani, M.; Shindo, H. In vivo structural analysis of subchondral trabecular bone in osteoarthritis of the hip using multi-detector row CT. Osteoarthr. Cartil. 2011, 19, 180–185.
- Jaiprakash, A.; Prasadam, I.; Feng, J.Q.; Liu, Y.; Crawford, R.; Xiao, Y. Phenotypic characterization of osteoarthritic osteocytes from the sclerotic zones: A possible pathological role in subchondral bone sclerosis. Int. J. Biol. Sci. 2012, 8, 406–417.
- Lin, C.; Jiang, X.; Dai, Z.; Guo, X.; Weng, T.; Wang, J.; Li, Y.; Feng, G.; Gao, X.; He, L. Sclerostin mediates bone response to mechanical unloading through antagonizing Wnt/β-Catenin signaling. J. Bone Miner. Res. 2009, 24, 1651–1661.
- Tu, X.; Rhee, Y.; Condon, K.W.; Bivi, N.; Allen, M.R.; Dwyer, D.; Stolina, M.; Turner, C.H.; Robling, A.G.; Plotkin, L.I.; et al. Sost downregulation and local Wnt signaling are required for the osteogenic response to mechanical loading. Bone 2012, 50, 209–217.
- Jia, H.; Ma, X.; Wei, Y.; Tong, W.; Tower, R.J.; Chandra, A.; Wang, L.; Sun, Z.; Yang, Z.; Badar, F.; et al. Loading-Induced reduction in sclerostin as a mechanism of subchondral bone plate sclerosis in mouse knee joints during late-stage osteoarthritis. Arthritis Rheumatol. 2018, 70, 230–241.
- Ko, F.C.; Dragomir, C.; Plumb, D.A.; Goldring, S.R.; Wright, T.M.; Goldring, M.B.; van der Meulen, M.C.H. In vivo cyclic compression causes cartilage degeneration and subchondral bone changes in mouse tibiae. Arthritis Rheumatol. 2013, 65, 1569–1578.
- Bonnet, N.; Garnero, P.; Ferrari, S. Periostin action in bone. Mol. Cell. Endocrinol. 2016, 432, 75–82.
- Tajika, Y.; Moue, T.; Ishikawa, S.; Asano, K.; Okumo, T.; Takagi, H.; Hisamitsu, T. Influence of periostin on synoviocytes in knee osteoarthritis. In Vivo 2017, 31, 69–77.
- Chijimatsu, R.; Kunugiza, Y.; Taniyama, Y.; Nakamura, N.; Tomita, T.; Yoshikawa, H. Expression and pathological effects of periostin in human osteoarthritis cartilage. BMC Musculoskelet. Disord. 2015, 16, 215.
- Zhang, R.; Fang, H.; Chen, Y.; Shen, J.; Lu, H.; Zeng, C.; Ren, J.; Zeng, H.; Li, Z.; Chen, S.; et al. Gene expression analyses of subchondral bone in early experimental osteoarthritis by microarray. PLoS ONE 2012, 7, e32356.
- Finnilä, M.A.J.; Thevenot, J.; Aho, O.-M.; Tiitu, V.; Rautiainen, J.; Kauppinen, S.; Nieminen, M.T.; Pritzker, K.; Valkealahti, M.; Lehenkari, P.; et al. Association between subchondral bone structure and osteoarthritis histopathological grade. J. Orthop. Res. 2017, 35, 785–792.
- Ding, M.; Odgaard, A.; Danielsen, C.C.; Hvid, I. Mutual associations among microstructural, physical and mechanical properties of human cancellous bone. J. Bone Jt. Surg. Br. 2002, 84, 900–907.
- Wang, T.; Wen, C.Y.; Yan, C.H.; Lu, W.W.; Chiu, K.Y. Spatial and temporal changes of subchondral bone proceed to microscopic articular cartilage degeneration in guinea pigs with spontaneous osteoarthritis. Osteoarthr. Cartil. 2013, 21, 574–581.
- Conway, S.J.; Izuhara, K.; Kudo, Y.; Litvin, J.; Markwald, R.; Ouyang, G.; Arron, J.R.; Holweg, C.T.; Kudo, A. The role of periostin in tissue remodeling across health and disease. Cell. Mol. Life. Sci. 2014, 71, 1279–1288.
- Spatz, J.M.; Ellman, R.; Cloutier, A.M.; Louis, L.; van Vliet, M.; Suva, L.J.; Dwyer, D.; Stolina, M.; Ke, H.Z.; Bouxsein, M.L. Sclerostin antibody inhibits skeletal deterioration due to reduced mechanical loading. J. Bone Miner. Res. 2013, 28, 865–874.
- Gingery, A.; Subramaniam, M.; Pitel, K.S.; Li, X.; Ke, H.Z.; Turner, R.T.; Iwaniec, U.T.; Hawse, J.R. Sclerostin antibody treatment rescues the osteopenic bone phenotype of TGFβ inducible early gene-1 knockout female mice. J. Cell Physiol. 2020, 235, 5679–5688.
- Mazur, C.M.; Woo, J.J.; Yee, C.S.; Fields, A.J.; Acevedo, C.; Bailey, K.N.; Kaya, S.; Fowler, T.W.; Lotz, J.C.; Dang, A.; et al. Osteocyte dysfunction promotes osteoarthritis through MMP13-dependent suppression of subchondral bone homeostasis. Bone Res. 2019, 7, 34.
- Rowe, P.S.N. Regulation of bone-renal mineral and energy metabolism: The PHEX, FGF23, DMP1, MEPE ASARM pathway. Crit. Rev. Eukaryot. Gene Expr. 2012, 22, 61–86.
- Zhen, G.; Wen, C.; Jia, X.; Li, Y.; Crane, J.L.; Mears, S.C.; Askin, F.B.; Frassica, F.J.; Chang, W.; Yao, J.; et al. Inhibition of TGF-β signaling in mesenchymal stem cells of subchondral bone attenuates osteoarthritis. Nat. Med. 2013, 19, 704–712.
- Ling, Y.; Rios, H.F.; Myers, E.R.; Lu, Y.; Feng, J.Q.; Boskey, A.L. DMP1 depletion decreases bone mineralization in vivo: An FTIR imaging analysis. J. Bone Miner. Res. 2005, 20, 2169–2177.
- Baranyay, F.J.; Wang, Y.; Wluka, A.E.; English, D.R.; Giles, G.G.; Sullivan, R.O.; Cicuttini, F.M. Association of bone marrow lesions with knee structures and risk factors for bone marrow lesions in the knees of clinically healthy, community-based adults. Semin. Arthritis Rheum. 2007, 37, 112–118.
- Davies-Tuck, M.L.; Wluka, A.E.; Forbes, A.; Wang, Y.; English, D.R.; Giles, G.G.; O’Sullivan, R.; Cicuttini, F.M. Development of bone marrow lesions is associated with adverse effects on knee cartilage while resolution is associated with improvement--a potential target for prevention of knee osteoarthritis: A longitudinal study. Arthritis Res. Ther. 2010, 12, R10.
- Dore, D.; Martens, A.; Quinn, S.; Ding, C.; Winzenberg, T.; Zhai, G.; Pelletier, J.-P.; Martel-Pelletier, J.; Abram, F.; Cicuttini, F.; et al. Bone marrow lesions predict site-specific cartilage defect development and volume loss: A prospective study in older adults. Arthritis Res. Ther. 2010, 12, R222.
- Kornaat, P.R.; Kloppenburg, M.; Sharma, R.; Botha-Scheepers, S.A.; Le Graverand, M.-P.H.; Coene, L.N.J.E.M.; Bloem, J.L.; Watt, I. Bone marrow edema-like lesions change in volume in the majority of patients with osteoarthritis; associations with clinical features. Eur. Radiol. 2007, 17, 3073–3078.
- Narayanan, K.; Ramachandran, A.; Hao, J.; He, G.; Park, K.W.; Cho, M.; George, A. Dual functional roles of dentin matrix protein 1. Implications in biomineralization and gene transcription by activation of intracellular Ca2+ store. J. Biol. Chem. 2003, 278, 17500–17508.
- Zhou, Y.; Lin, J.; Shao, J.; Zuo, Q.; Wang, S.; Wolff, A.; Nguyen, D.T.; Rintoul, L.; Du, Z.; Gu, Y.; et al. Aberrant activation of Wnt signaling pathway altered osteocyte mineralization. Bone 2019, 127, 324–333.
- Gluhak-Heinrich, J.; Ye, L.; Bonewald, L.F.; Feng, J.Q.; MacDougall, M.; Harris, S.E.; Pavlin, D. Mechanical loading stimulates dentin matrix protein 1 (DMP1) expression in osteocytes in vivo. J. Bone Miner. Res. 2003, 18, 807–817.
- Ding, M.; Odgaard, A.; Hvid, I. Changes in the three-dimensional microstructure of human tibial cancellous bone in early osteoarthritis. J. Bone Jt. Surg. Br. 2003, 85, 906–912.
- Brown, S.J.; Pollintine, P.; Powell, D.E.; Davie, M.W.J.; Sharp, C.A. Regional differences in mechanical and material properties of femoral head cancellous bone in health and osteoarthritis. Calcif. Tissue Int. 2002, 71, 227–234.
- Ding, C.; Cicuttini, F.; Jones, G. Tibial subchondral bone size and knee cartilage defects: Relevance to knee osteoarthritis. Osteoarthr. Cartil. 2007, 15, 479–486.
- Zhang, M.; Xuan, S.; Bouxsein, M.L.; von Stechow, D.; Akeno, N.; Faugere, M.C.; Malluche, H.; Zhao, G.; Rosen, C.J.; Efstratiadis, A.; et al. Osteoblast-specific knockout of the insulin-like growth factor (IGF) receptor gene reveals an essential role of IGF signaling in bone matrix mineralization. J. Biol. Chem. 2002, 277, 44005–44012.
- Cooper, K.L.; Oh, S.; Sung, Y.; Dasari, R.R.; Kirschner, M.W.; Tabin, C.J. Multiple phases of chondrocyte enlargement underlie differences in skeletal proportions. Nature 2013, 495, 375–378.
- Sheng, M.H.; Lau, K.H.; Baylink, D.J. Role of osteocyte-derived insulin-like growth factor I in developmental growth, modeling, remodeling, and regeneration of the bone. J. Bone Metab. 2014, 21, 41–54.
- Tian, F.; Wang, Y.; Bikle, D.D. IGF-1 signaling mediated cell-specific skeletal mechano-transduction. J. Orthop. Res. 2018, 36, 576–583.
- Chubinskaya, S.; Hakimiyan, A.; Pacione, C.; Yanke, A.; Rappoport, L.; Aigner, T.; Rueger, D.C.; Loeser, R.F. Synergistic effect of IGF-1 and OP-1 on matrix formation by normal and OA chondrocytes cultured in alginate beads. Osteoarthr. Cartil. 2007, 15, 421–430.
- Loeser, R.F. Molecular mechanisms of cartilage destruction: Mechanics, inflammatory mediators, and aging collide. Arthritis Rheumatol. 2006, 54, 1357–1360.
- Rim, Y.A.; Nam, Y.; Ju, J.H. The role of chondrocyte hypertrophy and senescence in osteoarthritis initiation and progression. Int. J. Mol. Sci. 2020, 21, 2358.
- Sanchez-Adams, J.; Leddy, H.A.; McNulty, A.L.; O’Conor, C.J.; Guilak, F. The mechanobiology of articular cartilage: Bearing the burden of osteoarthritis. Curr. Rheumatol. Rep. 2014, 16, 451.
- Plumb, M.S.; Treon, K.; Aspden, R.M. Competing regulation of matrix biosynthesis by mechanical and IGF-1 signalling in elderly human articular cartilage in vitro. Biochim. Biophys. Acta 2006, 1760, 762–767.
- Chen, Y.; Hu, Y.; Yu, Y.E.; Zhang, X.; Watts, T.; Zhou, B.; Wang, J.; Wang, T.; Zhao, W.; Chiu, K.Y.; et al. Subchondral trabecular rod loss and plate thickening in the development of osteoarthritis. J. Bone Miner. Res. 2018, 33, 316–327.
- Muraoka, T.; Hagino, H.; Okano, T.; Enokida, M.; Teshima, R. Role of subchondral bone in osteoarthritis development: A comparative study of two strains of guinea pigs with and without spontaneously occurring osteoarthritis. Arthritis Rheumatol. 2007, 56, 3366–3374.
- Tomiyama, T.; Fukuda, K.; Yamazaki, K.; Hashimoto, K.; Ueda, H.; Mori, S.; Hamanishi, C. Cyclic compression loaded on cartilage explants enhances the production of reactive oxygen species. J. Rheumatol. 2007, 34, 556–562.
- Wolff, K.J.; Ramakrishnan, P.S.; Brouillette, M.J.; Journot, B.J.; McKinley, T.O.; Buckwalter, J.A.; Martin, J.A. Mechanical stress and ATP synthesis are coupled by mitochondrial oxidants in articular cartilage. J. Orthop. Res. 2013, 31, 191–196.
More