Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Catherine Yang and Version 1 by Rita Moretti.
Homocysteine (Hcy) is a sulfur-containing amino acid generated during methionine metabolism, accumulation of which may be caused by genetic defects or the deficit of vitamin B12 and folate.
- homocysteine
- SVD
- neurodegeneration
- vascular dementia
- endothelium damage
1. Introduction
1. Homocysteine and Brain
2. Homocysteine and Brain
Hcy is a sulfur-containing intermediary amino acid [132][1], recycled via the remethylation pathway or converted into cysteine via the trans-sulfuration pathway [4][2].
The methionine synthesis occurs when there is a reduction of 5,10-methylenetetrahydrofolate to 5-methyltetrahydrofolate (5-methylTHF) [133,134,135][3][4][5]. In remethylation, Hcy acquires a methyl group from N-5-methyltetrahydrofolate or from betaine to form methionine. The reaction with N-5-methyltetrahydrofolate occurs in all tissues and is vitamin B12-dependent. In particular, methionine adenosyltransferase (MAT) catalyzes S-adenosylmethionine (AdoMet) (SAM), actively consuming Adenosyn triphosphate (ATP) [133,134][3][4]. SAM is the methyl group donor in numerous methylation reactions, a fundamental process for the protein, phospholipid, and biogenic amines synthesis [136,137,138,139,140,141][6][7][8][9][10][11]. Every reaction made by methyltransferases produces S-adenosylhomocysteine (AdoHcy) (SAH) [142,143,144,145][12][13][14][15]. The SAM to SAH ratio defines the cell’s methylation potential [146,147,148,149,150,151,152,153,154,155][16][17][18][19][20][21][22][23][24][25].
In the trans-sulfuration pathway, Hcy condenses with serine to form cystathionine. It is an irreversible reaction catalyzed by the pyridoxal-50-phosphate (PLP)-containing enzyme, cystathionine β-synthase. Cystathionine is hydrolyzed by a second PLP-containing enzyme, γ-cystathionase, to form cysteine and α-ketobutyrate [144][14]. Excess cysteine is oxidized to taurine or inorganic sulfates or is excreted in the urine [144][14]. Therefore, the trans-sulfuration pathway catabolizes excess homocysteine, which is not required for methyl transfer [144,151,152,153,154,155][14][21][22][23][24][25].
The intrinsic capacity to differentiate between the remethylation and trans-sulfuration pathways to adapt to different intake-methionine levels in the diet strongly implies the existence of a coordinate regulation between these two pathways [144][14]. SAM could act as an allosteric inhibitor of methylenetetrahydrofolate reductase (MTHFR). It could also play a role as an activator of cystathionine β-synthase, promoting the trans-sulfuration pathway (cystathionine synthesis) [144][14]. When the methionine supply is low, there is an elevated rate of N-5-methyltetrahydrofolate production. Thus, remethylation will be favored over trans-sulfuration because the concentration of SAM is too low to activate the cystathionine β-synthase enzyme [144][14]. Remethylation of Hcy to methionine (the methionine cycle) predominates over the catabolic degradation of Hcy (trans-sulfuration) because of the order of magnitude difference in Km between MS and CBS [155,156,157,158,159,160][25][26][27][28][29][30].
The methylation reactions are necessary for the brain, SAM being the sole methyl group donor in numerous methylation reactions involving proteins, phospholipids, and biogenic amines, and packaging many phospholipids, i.e., polyunsaturated phosphatidylcholines (PC).
Hyper-homocysteinemia (HHcy) is defined as levels > 15 mol/L, levels between 15 and 30 are considered moderate HHcy, levels at 30–100 micro-mol/L are considered intermediate/severe HHCy, and levels above 100 micro-mol/L are considered as severe (often fatal) HHcy [156,157][26][27]. Hcy levels are inversely related to food supplements, principally folate and vitamin B12 [158[28][29][30],159,160], and directly related to smoking, alcohol, physical apathy [161[31][32],162], and aging [163][33]. In vitro studies that explored the correlation between Hcy and inflammation, neurodegeneration, atherosclerosis, and oxidative damage have been inconclusive. Similarly, in vivo trials failed to demonstrate a real benefit in clinical conditions when Hcy is abated by vitamin B12 or B9 supplementation [164][34].
Genetic causes of severe HHcy linked to a deficiency of CBS or other alterations of remethylation and trans-sulfuration pathways have been reported in neural tube defects and blood–brain barrier alterations [165,166,167,168,169][35][36][37][38][39]. Clinical and experimental works demonstrate that HHcy decreases the cell’s methylation potential, modifying the SAM/SAH ratio [170[40][41][42],171,172], and this is the primary determinant for a generalized DNA hypomethylation associated with an excess of oxidative stress [144,170][14][40].
Homocysteine accumulation could interfere with endothelium dysregulation, favor oxidative damage, and promote neuroinflammation and neurodegenerative processes [163,171,172,173,174,175][33][41][42][43][44][45]. All these processes occur in SVD; nevertheless, few studies directly focus on HHcy and SVD. Our review attempts to shed some light on the three principal mechanisms of HHcy-induced damage, trying to focus on SVD (Figure 31).
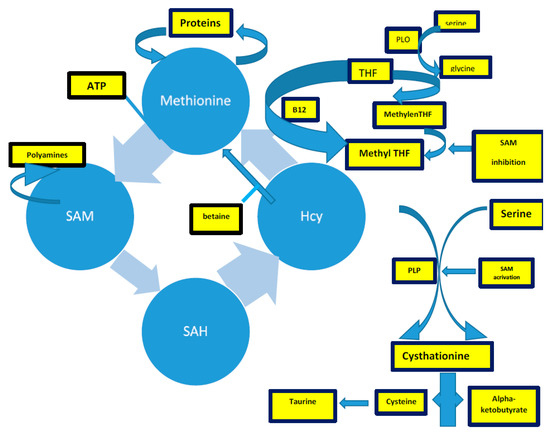
Figure 31. The complex of Hcy production, described in the text. Acronyms: SAM: s-adenosylmethionine; THF: tetrahydrofolate; PLP: pyridoxal-5-phosphate.
32. Homocysteine and Neurodegeneration
HHcy is linked to neurodegeneration, starting from the well-known relationship between its elevation during aging. Many in vivo and in vitro studies showed that HHcy favors the Abeta1–40 deposition in AD [174][44], mediated by an Hcy-induced upregulation of the Endoplasmic Reticulum Protein (HERP). HERP favors the c-secretase enzyme activity and the consequent increment of the intra- and extra-cellular accumulation of Abeta1–40 and Abeta 42 [175,176,177,178][45][46][47][48].
Hcy is strongly related to neurodegenerative/neuroinflammation conditions by the homocysteinylation process. Homocysteinylation leads to protein damage, i.e., protein denaturation, enzyme inactivation, inflammatory activities, and amyloid-oligomers deposition [179,180,181,182,183,184,185][49][50][51][52][53][54][55]. Under normal metabolic conditions, the cellular synthesis of Hcy thiolactone is rather low because intracellular concentrations of Hcy are relatively low [186][56]. If Hcy levels are increased because of a reduction in transmethylation and/or trans-sulfuration, Hcy thiolactone synthesis is enhanced—it could be as much as 60% of the metabolized Hcy [186][56]. Hcy can be linked to a protein via an isopeptide bond to lysine (Lys) residues (N-Hcy-protein) [187,188,189][57][58][59] or via a disulfide bond to Cys residues (S-Hcy-protein) [190,191,192,193][60][61][62][63]. N-homocysteinylation is an emerging post-translational protein modification that impairs or alters the protein’s structure/function and causes protein damage [194][64]. There are two limiting processes of the N-homocysteinylation: the quantity of cyclic Hcy-thiolactone (dependent on HHcy) and the number of lysine residues encountered [195,196,197][65][66][67]. The most evident result of the general homocysteinylation process is protein aggregation and virtual protein misfolding. Thus, Hcy-thiolactone induces apoptosis directly in endothelial cell cultures in in vitro and in vivo models [195][65].
Hcy is also linked to neurodegenerative pathology by influencing tau phosphorylation. As previously described [4[2][68],9], tau protein has many functions: the correct assembly of microtubules, directing, therefore, the axonal micronutrients transport toward the neuronal soma. The active form of tau needs constant dephosphorylation mediated by methyltransferase systems (the so-called PPM1 and PPM2A), and the methylation occurs through SAM-dependent reactions [198,199,200,201,202][69][70][71][72][73]. Tau hyperphosphorylation has two direct consequences: (1) the disaggregation of microtubules, which leads to an inhibition of axonal transport, and (2) a neuronal death, together with a deposition of damaged microtubules, which forms the so-called tau depositions, or neurofibrillary tangles [203,204,205][74][75][76]. These phenomena have always been associated with degenerative conditions (AD, frontal Pick complex, etc.), but they have also been demonstrated in neuroblastoma cultured cells when the culture medium is depleted by folate, and an increase of P-tau by 66% occurs [206][77].
HHcy has an intrinsic toxic property [4,9,207][2][68][78] as it acts as an agonist of NMDA (N-methyl D-Aspartate) receptors [208,209,210,211][79][80][81][82] depending on glycine concentration. Hcy acts as a partial antagonist of the NMDA receptors [4,162,171,207[2][32][41][78][79],208], but when the glycine concentration is increased (like in the brain ischemia, in vasospasms, i.e., in prolonged migraine aura attack), even low doses of Hcy could act as an agonist of NMDA channels [212[83][84],213], inducing an enhancement of calcium flows [213][84]. HHCy promotes an extracellular signal-regulated kinase activity in the hippocampus, regulated or blocked by three glutamate receptor antagonists (NMDA, not-NMDA, and metabotropic receptors) [154,214][24][85]. It has been suggested that Hcy could directly activate group I metabotropic glutamate receptors, favoring calcium influx currents [212][83].
Collectively, HHcy exerts essential alteration in the SVD pattern. HHcy induces an increase of Abeta 1–40 toxicity on the smooth muscle cells of the brain’s small arteries, where cerebral amyloid depositions occur, transforming the event into cerebral amyloid angiopathy (CAA), a constant finding in overt SVD condition [4,9,215,216,217][2][68][86][87][88]. Moreover, the HHcy condition enhances the m-RNA (Messanger-RNA) production of the C-reactive protein (CRP), over-expressing the NR1 subunit of NMDA receptor expression [4,218][2][89]. HHcy enhances the signal pathway cascade, mediated by CRP hyperproduction, mediated by NMDA-ROS-erk1/2/p38-NFK-Beta (NFK = Nuclear Kappa Factor-Beta), which occurs in the smooth muscle cells’ brain small arteries [218][89]. Homocysteinilation promotes apoptosis [195][65], endothelium alterations, protein misfolding, and protein aggregation. In fact, the multiple lysine-rich proteins are fibrinogen [196[66][90],219], high-density lipoprotein [220][91], lysine oxidase [221][92], and cytochrome c [197][67], and all of them homocysteinylate, aggregate [195][65], and lead to a general pro-thrombotic condition [196[66][91][92][93],220,221,222], enhanced coagulation [223][94], and reduced fibrinolysis [224,225][95][96].
References
- Smith, A.D.; Refsum, H. Homocysteine, B vitamins, and cognitive impairment. Annu. Rev. Nutr. 2016, 36, 211–239.
- Moretti, R.; Caruso, P. The Controversial Role of Homocysteine in Neurology: From Labs to Clinical Practice. Int. J. Mol. Sci. 2019, 20, 231.
- Blom, H.J.; Smulders, Y. Overview of homocysteine and folate metabolism. With special references to cardiovascular disease and neural tube defects. J. Inherit. Metab. Dis. 2011, 34, 75–81.
- Loscalzo, J.; Handy, D.E. Epigenetic modifications: Basic mechanisms and role in cardiovascular disease. 2013 Grover Conference Series. Pulm. Circ. 2014, 482, 169–174.
- Miles, L.; Allen, E.; Mills, K.; Clarke, R.; Uauy, R.; Dangour, A.D. Vitamin B12 status and neurologic function in older people: A cross-sectional analysis of baseline trial data from the Older People and Enhanced Neurological Function (OPEN) study. Am. J. Clin. Nutr. 2016, 104, 790–796.
- Obeid, R.; Herrmann, W. Mechanisms of homocysteine neurotoxicity in neurodegenerative diseases with special reference to dementia. FEBS Lett. 2006, 580, 2994–3005.
- Price, B.R.; Wilcock, D.M.; Weekman, E.M. Hyeprhomocysteinemia as a risk factor for vascular contributions to cognitive impairment and dementia. Front. Aging Neurosci. 2018, 10, 305.
- Mudd, S.H.; Cantoni, G.L. Activation of methionine for transmethylation. III. The methionine-activating enzyme of Bakers’ yeast. J. Biol. Chem. 1958, 231, 481–492.
- Mato, J.M.; Alvarez, L.; Ortiz, P.; Pajares, M.A. S-adenosylmethionine synthesis: Molecular mechanisms and clinical implications. Pharmacol. Ther. 1997, 73, 265–280.
- Taha, S.; Azzi, A.; Ozer, N.K. Homocysteine induces DNA synthesis and proliferation of vascular smooth muscle cells by a hydrogen peroxide-independent mechanism. Antioxid. Redox Signal. 1999, 1, 365–369.
- Robinson, J.L.; McBreairty, L.E.; Randell, E.W.; Harding, S.V.; Bartlett, R.K.; Brunton, J.A.; Bertolo, R.F. Betaine or folate can equally furnish remethylation to methionine and increase transmethylation in methionine-restricted neonates. J. Nutr. Biochem. 2018, 59, 129–135.
- Kotb, M.; Mudd, S.H.; Mato, J.M. Consensus nomenclature for the mammalian methionine adenosyltransferase genes and gene products. Trends Genet. 1997, 13, 51–52.
- Smulders, Y.M.; Blom, H.J. The homocysteine controversy. J. Inherit. Metab. Dis. 2011, 34, 93–99.
- Selhub, J. Homocysteine metabolism. Annu. Rev. Nutr. 1999, 19, 217–246.
- Parnetti, L.; Bottiglieri, T.; Lowenthal, D. Role of homocysteine in age-related vascular and non-vascular diseases. Aging Clin. Exp. Res. 1997, 9, 241–257.
- Handy, D.E.; Castro, R.; Loscalzo, J. Epigenetic modifications: Basic mechanisms and role in cardiovascular disease. Circulation 2011, 123, 2145–2156.
- Enk, C.; Hougaard, K.; Hippe, E. Reversible dementia and neuropathy associated with folate deficiency 16 years after partial gastrectomy. Scand. J. Haematol. 1980, 25, 63–66.
- Bottiglieri, T. Ademetionine (S-adenosylmethionine) neuropharmacology: Implications for drug therapies in psychiatric and neurological disorders. Expert Opin. Investig. Drugs 1997, 6, 417–426.
- Weir, D.G.; Keating, S.; Molloy, A. Methylation deficiency causes vitamin B12-associated neuropathy in the pig. J. Neurochem. 1988, 51, 1949–1952.
- Surtees, R.; Leonard, J.; Austin, S. Association of demyelination with deficiency of cerebrospinal-fluid S-adenosylmethionine in inborn errors of methyl-transfer pathway. Lancet 1991, 338, 1550–1554.
- Pennypacker, L.C.; Allen, R.H.; Kelly, J.P. High prevalence of cobalamin deficiency in elderly outpatients. J. Am. Geriatr. Soc. 1992, 40, 1197–1204.
- McKeever, M.P.; Weir, D.G.; Molloy, A.; Scott, J.M. Betaine-homocysteine methyltransferase: Organ distribution in man, pig and rat and subcellular distribution in the rat. Clin. Sci. 1991, 81, 551–556.
- Leclerc, D.; Wilson, A.; Dumas, R. Cloning and mapping of a cDNA for methionine synthase reductase, a flavoprotein defective in patients with homocystinuria. Proc. Natl. Acad. Sci. USA 1998, 95, 3059–3064.
- Sunden, S.L.; Renduchintala, M.S.; Park, E.I.; Miklasz, S.D.; Garrow, T.A. Betaine-homocysteine methyltransferase expression in porcine and human tissues and chromosomal localization of the human gene. Arch. Biochem. Biophys. 1997, 345, 171–174.
- Quéré, I.; Paul, V.; Rouillac, C. Spatial and temporal expression of the cystathionine beta-synthase gene during early human development. Biochem. Biophys. Res. Commun. 1999, 254, 127–137.
- Pietrzik, K.; Bronstrup, A. Vitamins B12, B6 and folate as determinants of homocysteine concentration in the healthy population. Eur. J. Pediatr. 1998, 157 (Suppl. S2), S135–S138.
- Huang, Y.C.; Chang, S.J.; Chiu, Y.T.; Chang, H.H.; Cheng, C.H. The status of plasma homocysteine and related B-vitamins in healthy young vegetarians and nonvegetarians. Eur. J. Nutr. 2003, 42, 84–90.
- Kulkarni, K.; Richard, B.C. Lifestyle, homocysteine and the metabolic syndrome. Metab. Syndr. Relat. Disord. 2003, 1, 141–147.
- Ansari, R.; Mahta, A.; Mallack, E.; Luo, J.J. Hyperhomocysteinemia and neurologic disorders: A review. J. Clin. Neurol. 2014, 10, 281–288.
- Stea, T.H.; MAnsoor, M.A.; Wandel, M.; Uglem, S.; Frolich, W. Changes in predictors and status of homocysteine in young male adults after dietary intervention with vegetables, fruits and bread. Eur. J. Nutr. 2008, 47, 201–209.
- Pushpakumar, S.; Kundu, S.; Sen, U. Endothelial dysfunction: The link between homocysteine and hydrogen sulfide. Curr. Med. Chem. 2014, 21, 3662–3672.
- Moretti, R.; Dal Ben, M.; Gazzin, S.; Tiribelli, C. Homcysteine in neurology: From endothelium to neurodegeneration. Curr. Nutr. Food Sci. 2017, 13, 163–175.
- Surtees, R.; Bowron, A.; Leonard, J. Cerebrospinal fluid and plasma total homocysteine and related metabolites in children with cystathionine beta-synthase deficiency: The effect of treatment. Pediatr. Res. 1997, 42, 577–582.
- Afman, L.A.; Blom, H.J.; Drittij, M.J.; Brouns, M.R.; van Straaten, H.W. Inhibition of transmethylation disturbs neurulation in chick embryos. Brain Res. Dev. Brain Res. 2005, 158, 59–65.
- Kamath, A.F.; Chauhan, A.K.; Kisucka, J. Elevated levels of homocysteine compromise blood-brain barrier integrity in mice. Blood 2006, 107, 591–593.
- Troen, A.M. The central nervous system in animal models of hyperhomocysteinemia. Prog. NeuroPsychopharmacol. Biol. Psychiatry 2005, 29, 1140–1151.
- Algaidi, S.A.; Christie, L.A.; Jenkinson, A.M. Long-term homocysteine exposure induces alterations in spatial learning, hippocampal signalling and synaptic plasticity. Exp. Neurol. 2006, 197, 8–21.
- Ganguly, P.; Alam, S.F. Role of homocysteine in the development of cardiovascular disease. Nutr. J. 2015, 14, 6.
- Sultan, M.O.; Farooque, U.; Javed, R.; Khan, M.I.; Karimi, S.; Abdul Sattar, R.; Cheema, O. Correlation of Homocysteine Level and Age in Patients with Ischemic Stroke. Cureus 2020, 12, e7785.
- Moretti, R.; Peinkhofer, C. B Vitamins and Fatty Acids: What Do They Share with Small Vessel Disease-Related Dementia? Int. J. Mol. Sci. 2019, 20, 5797.
- Moretti, R. Homocysteine: New Aspects of an Ancient Enigma. Cardiology 2019, 144, 36–39.
- Piao, X.; Wu, G.; Yang, P.; Shen, J.; De, A.; Wu, J.; Qu, Q. Association between Homocysteine and Cerebral Small Vessel Disease: A Meta-Analysis. J. Stroke Cerebrovasc. Dis. 2018, 27, 2423–2430.
- Rutten-Jacobs, L.C.A.; Traylor, M.; Adib-Samii, P.; Thijs, V.; Sudlow, C.; Rothwell, P.M.; Boncoraglio, G.; Dichgans, M.; Meschia, J.; Maguire, J.; et al. Association of MTHFR C677T Genotype With Ischemic Stroke Is Confined to Cerebral Small Vessel Disease Subtype. Stroke 2016, 47, 646–651.
- Irizarry, M.C.; Gurol, M.E.; Raju, S. Association of homocysteine with plasma amyloid beta protein in aging and neurodegenerative disease. Neurology 2005, 65, 1402–1408.
- Hasegawa, T.; Ukai, W.; Jo, D.-G. Homocysteic acid induces intraneuronal accumulation of neurotoxic Abeta42, implications for the pathogenesis of Alzheimer’s disease. J. Neurosci. Res. 2005, 80, 869–876.
- Morris, M.S. Homocysteine and Alzheimer’s disease. Lancet Neurol. 2003, 2, 425–428.
- Kruman, I.I.; Kumaravel, T.S.; Lohani, A. Folic acid deficiency and homocysteine impair DNA repair in hippocampal neurons and sensitize them to amyloid toxicity in experimental models of Alzheimer’s disease. J. Neurosci. 2002, 22, 1752–1762.
- Sai, X.; Kawamura, Y.; Kokame, K. Endoplasmic reticulum stress-inducible protein, Herp, enhances presenilin-mediated generation of amyloid beta-protein. J. Biol. Chem. 2002, 277, 12915–12920.
- Selkoe, D.J. Presenilin, Notch, and the genesis and treatment of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2001, 98, 11039–11041.
- Scarpa, S.; Fuso, A.; D’Anselmi, F.; Cavallaro, R.A. Presenilin 1 gene silencing by S-adenosylmethionine: A treatment for Alzheimer disease? FEBS Lett. 2003, 541, 145–148.
- Baernstein, H.D. A modification of the method for determining methionine in proteins. J. Biol. Chem. 1934, 106, 451–456.
- Jakubowski, H.; Fersht, A. Alternative pathways of rejection of noncognate amino acids by aminoacyl-tRNA synthetases. Nucleic Acids Res. 1981, 9, 3105–3117.
- Jakubowski, H. Proofreading in vivo: Editing of homocysteine by methionyl-tRNA synthetase in Escherichia coli. Proc. Natl. Acad. Sci. USA 1990, 87, 4504–4508.
- Jakubowski, H. Metabolism of homocysteine thiolactone in human cell cultures: Possible mechanism for pathological consequences of elevated homocysteine levels. J. Biol. Chem. 1997, 272, 1935–1942.
- Sharma, G.S.; Kumar, T.; Dar, T.A.; Singh, L.R. Protein-N-Homocysteinylation: Form cellular toxicity to neurodegeneration. Biochim. Et Biophys. Acta 2015, 1850, 2239–2245.
- Jakubowski, H. Homocysteine Thiolactone: Metabolic Origin and Protein Homocysteinylation in Humans. J. Nutr. 2000, 130, 377S–381S.
- Jakubowski, H. Homocysteine is a protein amino acid in humans. Implications for homocysteine-linked disease. J. Biol. Chem. 2002, 277, 30425–30428.
- Jakubowski, H. Protein homocysteinylation: Possible mechanism underlying pathological consequences of elevated homocysteine levels. FASEB J. 1999, 13, 2277–2283.
- Sikora, M.; Marczak, Ł.; Kubalska, J.; Graban, A.; Jakubowski, H. Identification of N-homocysteinylation sites in plasma proteins. Amino Acids 2014, 46, 235–244.
- Jacovina, A.T.; Deora, A.B.; Ling, Q.; Broekman, M.J.; Almeida, D.; Greenberg, C.B.; Marcus, A.J.; Smith, J.D.; Hajjar, K.A. Homocysteine inhibits neoangiogenesis in mice through blockade of annexin A2-dependent fibrinolysis. J. Clin. Investig. 2009, 119, 3384–3394.
- Lim, A.; Sengupta, S.; McComb, M.E.; Théberge, R.; Wilson, W.G.; Costello, C.E.; Jacobsen, D.W. In vitro and in vivo interactions of homocysteine with human plasma transthyretin. J. Biol. Chem. 2003, 278, 49707–49713.
- Jakubowski, H. Homocysteine Modification in Protein Structure/Function and Human Disease. Physiol. Rev. 2019, 99, 555–604.
- Hortin, G.L.; Seam, N.; Hoehn, G.T. Bound homocysteine, cysteine, and cysteinylglycine distribution between albumin and globulins. Clin. Chem. 2006, 52, 2258–2264.
- Jakubowski, H. Homocysteine in Protein Structure/Function and Human Disease—Chemical Biology of Homocysteine-Containing Proteins; Springer: Vienna, Austria, 2013.
- Lai, W.K.; Kan, M.Y. Homocysteine-Induced Endothelial Dysfunction. Ann. Nutr. Metab. 2015, 67, 1–12.
- Perla-Kajan, J.; Twardowski, T.; Jakubowski, H. Mechanisms of homocysteine toxicity in humans. Amino Acids 2007, 32, 561–572.
- Frey, D.; Braun, O.; Briand, C.; Vasak, M.; Grutter, M.G. Structure of the mammalian NOS regulator dimethylarginine dimethylaminohydrolase: A basis for the design of specific inhibitors. Structure 2006, 14, 901–911.
- Caruso, P.; Signori, R.; Moretti, R. Small vessel disease to subcortical dementia: A dynamic model, which interfaces aging, cholinergic dysregulation and the neurovascular unit. Vasc. Health Risk Manag. 2019, 15, 259–281.
- Leulliot, N.; Quevillon-Cheruel, S.; Sorel, I. Structure of protein phosphatase methyltransferase 1 (PPM1), a leucine carboxyl methyltransferase involved in the regulation of protein phosphatase 2A activity. J. Biol. Chem. 2004, 279, 8351–8358.
- Ferreira, A.; Lu, Q.; Orecchio, L.; Kosik, K.S. Selective phosphorylation of adult tau isoforms in mature hippocampal neurons exposed to fibrillar A beta. Mol. Cell Neurosci. 1997, 9, 220–234.
- Wang, J.Z.; Gong, C.X.; Zaidi, T.; Grundke-Iqbal, I.; Iqbal, K. Dephosphorylation of Alzheimer paired helical filaments by protein phosphatase-2A and -2B. J. Biol. Chem. 1995, 270, 4854–4860.
- Vogelsberg-Ragaglia, V.; Schuck, T.; Trojanowski, J.Q.; Lee, V.M. PP2A mRNA expression is quantitatively decreased in Alzheimer’s disease hippocampus. Exp. Neurol. 2001, 168, 402–412.
- Sontag, E.; Hladik, C.; Montgomery, L. Downregulation of protein phosphatase 2A carboxyl methylation and methyltransferase may contribute to Alzheimer disease pathogenesis. J. Neuropathol. Exp. Neurol. 2004, 63, 1080–1091.
- Zhao, W.-Q.; Feng, C.; Alkon, D.L. Impairment of phosphatase 2A contributes to the prolonged MAP kinase phosphorylation in Alzheimer’s disease fibroblasts. Neurobiol. Dis. 2003, 14, 458–469.
- Vafai, S.B.; Stock, J.B. Protein phosphatase 2A methylation: A link between elevated plasma homocysteine and Alzheimer’s Disease. FEBS Lett. 2002, 518, 1–4.
- Tolstykh, T.; Lee, J.; Vafai, S.; Stock, J.B. Carboxyl methylation regulates phosphoprotein phosphatase 2A by controlling the association of regulatory B subunits. EMBO J. 2000, 19, 5682–5691.
- Ho, P.I.; Ashline, D.; Dhitavat, S. Folate deprivation induces neurodegeneration: Roles of oxidative stress and increased homocysteine. Neurobiol. Dis. 2003, 14, 32–42.
- Wuerthele, S.E.; Yasuda, R.P.; Freed, W.J.; Hoffer, B.J. The effect of local application of homocysteine on neuronal activity in the central nervous system of the rat. Life Sci. 1982, 31, 2683–2691.
- Lipton, S.A.; Kim, W.K.; Choi, Y.B. Neurotoxicity associated with dual actions of homocysteine at the N-methyl-D-aspartate receptor. Proc. Natl. Acad. Sci. USA 1997, 94, 5923–5928.
- Ito, S.; Provini, L.; Cherubini, E. L-homocysteic acid mediates synaptic excitation at NMDA receptors in the hippocampus. Neurosci. Lett. 1991, 124, 157–161.
- Klancnik, J.M.; Cuénod, M.; Gähwiler, B.H.; Jiang, Z.P.; Do, K.Q. Release of endogenous amino acids, including homocysteic acid and cysteine sulphinic acid, from rat hippocampal slices evoked by electrical stimulation of Schaffer collateral-commissural fibres. Neuroscience 1992, 49, 557–570.
- Kim, J.P.; Koh, J.Y.; Choi, D.W. L-homocysteate is a potent neurotoxin on cultured cortical neurons. Brain Res. 1987, 437, 103–110.
- Ziemiffska, E.; Stafiej, A.; Lazarewicz, J.W. Role of group I metabotropic glutamate receptors and NMDA receptors in homocysteine-evoked acute neurodegeneration of cultured cerebellar granule neurones. Neurochem. Int. 2003, 43, 481–492.
- Shi, Q.; Savage, J.E.; Hufeisen, S.J. L-homocysteine sulfinic acid and other acidic homocysteine derivatives are potent and selective metabotropic glutamate receptor agonists. J. Pharmacol. Exp. 2003, 305, 131–142.
- Robert, K.; Pagès, C.; Ledru, A. Regulation of extracellular signal-regulated kinase by homocysteine in hippocampus. Neuroscience 2005, 133, 925–935.
- De Lau, L.M.; Koudstaal, P.J.; van Meurs, J.B.; Uitterlinden, A.G.; Hofman, A.; Breteler, M.M. Methylenterahydrofolate reductase C677T genotype and PD. Annu. Neurol. 2005, 57, 927–930.
- Zhao, P.; Yang, J.F.; Liu, W.; Wang, Y.; Sun, Y.N.; Li, Q. Effects of entacapone on plasma homocysteine in Parkinson’s Disease patients on levodopoa. Zhongha Yi Xue Za Zhi 2013, 93, 512–515.
- Mok, S.S.; Turner, B.J.; Beyreuther, K. Toxicity of substrate-bound amyloid peptides on vascular smooth muscle cells is enhanced by homocysteine. Eur. J. Biochem. FEBS 2002, 269, 3014–3022.
- Pang, X.; Liu, J.; Zhao, J.; Mao, J.; Zhang, X.; Feng, L. Homocysteine induces the expression of C-reactive protein via NMDAr-ROS-MAPK-NF-KB signal pathway in rat vascular smooth muscle cells. Atherosclerosis 2014, 236, 73–81.
- Nelson, A.R.; Sweeney, M.D.; Sagare, A.P.; Zlokovic, B.V. Neurovascular dysfunction and neurodegeneration in dementia and Alzheimer’s disease. Biochim. Biophys. Acta 2016, 1862, 887–900.
- Jakubowski, H. The pathophysiological hypothesis of homocysteine thiolactone-mediated vascular disease. J. Physiol. Pharmacol. 2008, 59 (Suppl. S9), 155–167.
- Mercie, P.; Garnier, O.; Lascoste, L.; Renard, M.; Closse, C.; Durrieu, F.; Marit, G.; Boisseau, R.M.; Belloc, F. Homocysteine-thiolactone induces caspase-independent vascular endothelial cell death with apoptotic features. Apoptosis 2000, 5, 403–411.
- Dayal, S.; Wilson, K.M.; Leo, L.; Arning, E.; Bottiglieri, T.; Lentz, S.R. Enhanced susceptibility to arterial thrombosis in a murine model of hyperhomocysteinemia. Blood 2006, 108, 2237–2243.
- Undas, A.; Brozek, J.; Szczeklik, A. Homocysteine and thrombosis: From basic science to clinical evidence. Thromb. Haemost. 2005, 94, 907–915.
- Sauls, D.L.; Lockhart, E.; Warren, M.E.; Lenkowski, A.; Wilhelm, S.E.; Hoffman, M. Modification of fibrinogen by homocysteine thiolactone increases resistance to fibrinolysis: A potential mechanism of the thrombotic tendency in hyperhomocysteinemia. Biochemistry 2006, 45, 2480–2487.
- Tamura, Y.; Inoue, A.; Ijiri, Y.; Naemura, A.; Yamamoto, J. Short- and long-term treatment with folic acid suppresses thrombus formation in atherogenic mice in vivo. Pathophysiology 2014, 21, 169–175.
More