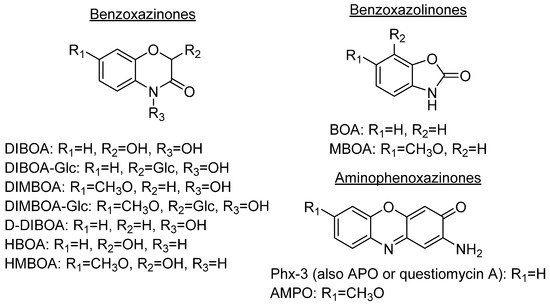
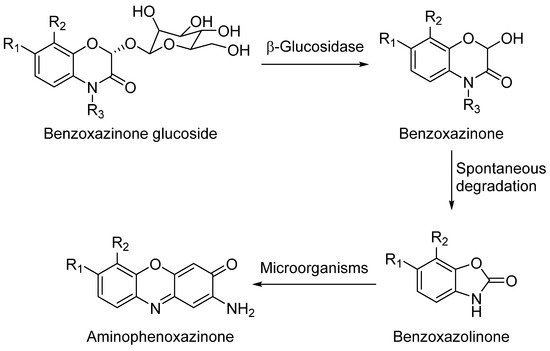
Aminophenoxazinones are degradation products resulting from the metabolism of different plant species, which comprise a family of natural products well known for their pharmacological activities. Aminophenoxazinones are tricyclic structures with double bonds in aromatic systems containing oxygen and nitrogen atom, which facilitates the development of synthetic derivatives to enhance the properties of these molecules. Aminophenoxazinones possess a number of promising properties like anticarcinogenic, antifungal, antiparasitic, antibacterial or antimicrobial activities.
- aminophenoxazinone
- drug
- Phx-3
- cytotoxicity
- cancer
1. Aminophenoxazinones as Degradation Products of Benzoxazinones
2. Anticancer Activity
2.1. Gastric and Colon Cancer
2.2. Glioblastoma
2.3. Melanoma
3. Activity of aminophenoxazinones on other cell lines
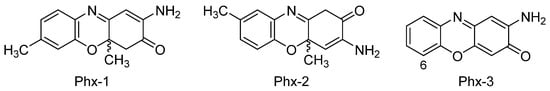
3.1 2-Aminophenoxazine-3-one (Phx-3)
First of all, we should highlight the study that Che et al. (2011) carried out to evaluate the cytotoxicity of Phx-3 on different types of cancer cells, such as MCF-7, A431, KCP-4, A549, KLM-1, MIA PaCa-2, ACHN, LoVo-1, U251MG and Y-79 lines [57]. In the case of ACHN (renal carcinoma), Liu et al. (2008) had already reported modest cytotoxic activity generated by Phx-3, as well as for a methylated and acetate derivative of this aminophenoxazinone [76]. Attending at IC50 values, the sensitivity of the mentioned cancer cell lines after 72 h of treatment with Phx-3 showed how almost all of them were vulnerable to Phx-3 at 10 µM (IC50 lower than 8 µM). KLM-1, Lovo-1 and Y79 lines were the exceptions (IC50 close to 20 µM). The normal cell lines HEL (embryonic pulmonary fibroblast) and HUVEC (umbilical cord) were also tested, being obtained high IC50 values (over 50 µM and 16 µM, respectively), which indicates that these cells are less sensitive to Phx-3 than cancer cells.
Regarding the pHi decrease, significant results were obtained for all cancer cell lines (reduction of 0.22-1.00 units at 20 µM, and 0.64-1.20 units at 100 µM), being comparable to those of the normal lines. The decrease was proved dose-dependent for MCF-7 (breast cancer) and A431 (skin cancer) cell lines for 30 min and ranging between 0 and 100 µM. Therefore, it can be concluded that the use of Phx-3 causes drastic acidification of cancer cells, which in turn induces their apoptosis [77]. Phx-3 would be a suitable drug for the treatment of cancer, as it causes drastic decreases in pHi (by more than 0.6), and induces apoptosis and cytotoxic effects on cancer cells without significant adverse effects.
MCF-7 and A431 lines were further evaluated to provide conclusions of the mechanism of action of Phx-3, attending at the reduction of the mitochondrial membrane potential (first and irreversible step towards apoptosis). The population of both MCF-7 and A431 lines, determined as their decrease in mitochondrial potential, increased as a direct function of both time and concentration of Phx-3. According to these data, and considering the reduction of the pHi value in both cell lines, it can be concluded that the apoptosis of MCF-7 and A431 cells could be preceded by the early acidification caused by Phx-3.
In other study, the cytotoxic and pro-apoptotic effects of Phx-3 on hepatocellular carcinoma dRLh-84 (rat) and HepG2 (human) cell lines, and the normal hepatocellular RLN-10 (rat) cell line have been studied [61]. Phx-3 reduced the number of viable cells in the three lines by a dose-dependent degree, being 2 µM enough to induce apoptosis by nuclear condensation and cell shrinkage. Moreover, Phx-3 combined with 2-deoxyglucose significantly enhanced apoptosis, but, on the other hand, some adverse effects were observed on normal liver cells. More recently, the combined treatment of Phx-3 with sorafenib was demonstrated to suppress the formation of hepatocellular carcinoma on in vivo studies. Phx-3 was reported as suppressor of the expression of GRP78, target protein directly related with different cancer cell lines, in HepG2 cells [78]. It must be noted that Phx-3 is named as questiomycin A in this last article.
The list of cancer lines with cell whose growth can be inhibited by Phx-3 may be completed with HeLa (cervical cancer, IC50 = 12.09 ± 3.29 µM) [35], U266 (myeloma), HL-60 (acute myeloid leukemia) and A549 (lung adenocarcinoma) lines[71]. The study of Moriya et al. (2011) was also focused on the pro-apoptotic transcription factor CHOP, being suggested that the regulation of its expression could represent a major target for treatments. So, in the case of U266, the activity of Phx-3 was enhanced by its combined application together with an inhibitor of NF-κB, a transcription factor related to the inhibition of CHOP.
3.2 Amino-4,4α-dihydro-4α,7-dimethyl-3H-phenoxazin-3-one (Phx-1)
Phx-1 (Figure 3) is an aminophenoxazinone with anticancer activity, obtained from the reaction of 2-amino-5-methylphenol with bovine hemoglobin [79]. The in vitro studies that have been conducted on this compound are similar to those previously described for Phx-3 [57].
Regarding the pHi reduction in MCF-7 and A431 cancer cell lines, similar or rather close results as those from Phx-3 were observed at the lowest concentrations (5-10 µM) were applied. However, Phx-1 proved to be significantly less active than Phx-3 at 50 µM, and concentration had to be increased up to 100 µM for any major differences to appear in the A431 line.
The study of Phx-1 on diverse cell lines (the same previously mentioned in the first paragraph of section 3.1), revealed that this compound achieves significant pHi reductions, but not as high as those attained by Phx-3. Thus, Phx-1 reached 0.01-0.16 at 20 µM and 0.11-0.59 at 100 µM, whereas Phx-3 achieved 0.22-1.00 at 20 µM and 0.64-1.20 at 100 µM. In both cases, the normal cell lines tested (HEL and HUVEC) suffered an equally significant decrease in pHi, comparable to that suffered by cancer cells.
The data obtained in relation to the cytotoxic effects of Phx-1, compared with those of Phx-3, revealed that the cytotoxicity generated by Phx-1 on all the tested cell lines was much lower (the most sensitive cancer line was MCF-7). These results are in agreement with the pHi changes undergone by the cells treated either Phx-1 or Phx-3 that have been summarized in the previous paragraph. This could represent a certain advantage regarding cancer treatment, since healthy cells would suffer a lesser damage.
Although the IC50 value of Phx-1 for Y-79 cells (retinoblastoma, eye cancer) is the highest, previous studies[80] showed in vivo antitumor effects in cells transplanted into mice whose strain suffers from a genetic mutation that causes the deterioration or lack of the thymus (organ where T cells mature). Phx-1 induced in vivo apoptosis to Y-79 in mice without any type of adverse effect even at high doses. These favorable signs make of Phx-1 a suitable candidate for the development of drugs for the treatment of retinoblastoma. In general, although Phx-1 is not highly effective, it could facilitate the induction of apoptosis and exert a cytotoxic effect on cancer cells.
It is worth highlighting that Phx-1 inhibited the proliferation and induced the apoptosis of diverse human leukemia cell lines (K562, HL-60 and HAL-01) in a dose-dependent manner. This study also proved that Phx-1 reduced in vivo the tumor growth rate in mice, whereas just few adverse effects were found on weight loss and white blood cell [81].
Once confirmed the ability of Phx-1 and Phx-3 to induce the apoptosis of diverse cancer cell lines, Tabuchi et al. (2011) studied the effects of these compounds to induce apoptotic cell death in human neutrophils, as this kind of cells are related with the mitochondrial depolarization and reduction of pHi. Both aminophenoxazines caused apoptosis or the loss of the morphology of neutrophils, while lymphocytes and monocytes did not undergo this process. These results would suggest that Phx-1 and Phx-3 are specific drugs to induce apoptotic cell death of neutrophils, being potential preventive anti-inflammatory drugs[58].
The antitumoral activity of Phx-1 and Phx-3 on NB-1 (neuroblastoma cell line) was demonstrated by another study. It was thereby confirmed that both aminophenoxazinones induced apoptosis and necrosis, being the IC50 value of Phx-3 much lower (0.5 µM vs. 20 µM)[82].
From the structural point of view, the differences in the bioactivity exhibited by Phx-1 and Phx-3 would be associated to the methyl group in Phx-1, even this premise is still to be confirmed. Molecular dynamics simulations could cast some light on the solubility of these compounds in cell membranes, which would allow to determine how much their structural differences affect cross-membrane processes and, thereby, explain the differences in their activity levels. In this line, certain recent studies have proven that the replacement of hydroxyl groups by fluorinated esters (in the eudesmanolide structure), improves the cytotoxicity of these compounds to cancer cells (HeLa, cervical cancer)[83].
3.3 Phenoxazine-indole conjugates
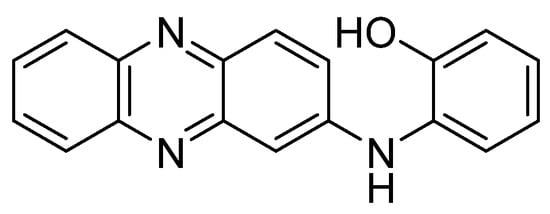
Recent studies have tested the activity by different derivatives with an indole group on different cancer cell lines. Indole groups are heterocyclic compounds formed by a benzene atom linked to a pyrrole compound, that have a pair of free electrons in the nitrogen atom of their aromatic ring. It is a fairly common component in perfumes, drug candidates and hormones (like melatonin)[84]. Some natural indole alkaloids, such as vincristine (Figure 5), has been accepted in USA by the Food and Drug Administration (FDA) as antitumor drugs[85].
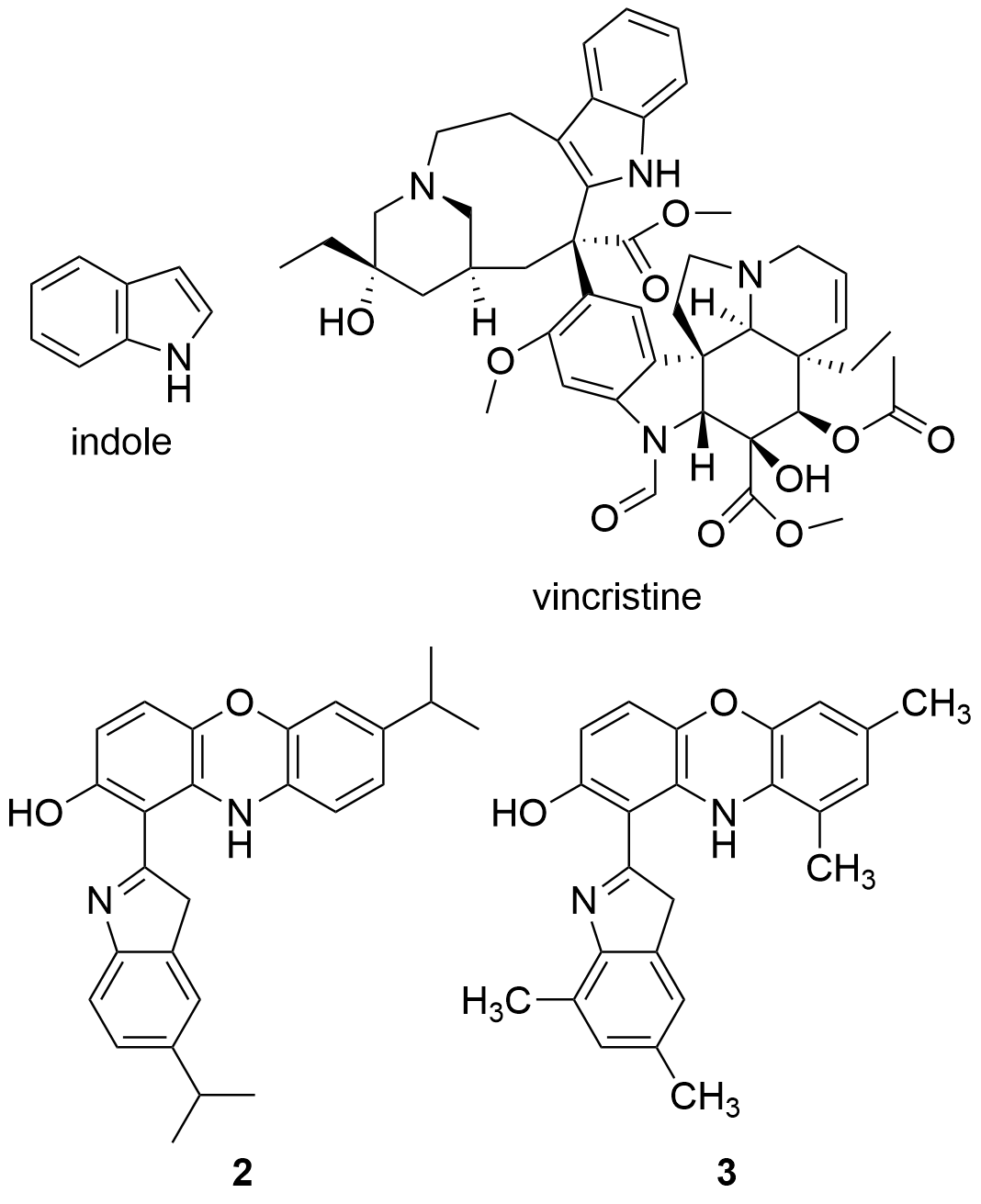
Figure 5. Structures of indole, the anticancer drug vincristine and the most active indole derivatives.
Taking into account the potential pharmacological effects of indoles, as well as those already recognized of phenoxazines and the synergistic effects exhibited by some pharmacophoric hybrids[86], Nunewar et al. (2020)[87] suggested that certain hybrids formed by two of these groups would possess some proliferative effect by operating as DNA exchangers. The general proposed mechanism consists of a first transfer of the molecule to the hydrophobic space between two adjacent DNA base pairs. As consequence, DNA would undergo conformational changes, in order for the molecule to accommodate itself between the base pairs. The resulting complex prevents DNA replication, which would lead to the death of the cell, being these results especially useful for the treatment of rapidly growing cancer cells.
Therefore, the cytotoxicity of certain indol derivatives was tested by Nunewar et al. (2020) on A549 (lung), MG-63 (bone), BT-474 (breast), Hep G2 (liver) and HCT116 (colon) human cancer cell lines, along with a normal line of lung epithelial tissue (L-132). All the derivatives tested showed remarkable IC50 values. The most inhibited cell line was A549, by the derivative 2 (IC50 = 3.71 ± 0.57 µM), characterized by two isopropyl substituents (Figure 5), and followed by compound 3 (IC50 = 4.43 ± 0.64 µM). In addition, 2 was the only derivative with the previously explained DNA intercalation capacity. These results confirm that hybrid compounds are a promising alternative for the search of new anticancer drugs.
3.4 Pyridophenoxazinone derivatives conjugated to L-lysine
Also in 2020 a number of studies were conducted on a series of pyridophenoxazinones (which possess intercalating capacity, and for generating free radicals that induce cell death by oxidative-stress) conjugated with the amino acid L-lysine, which were designed and synthesized with the aim of developing novel drug compounds with anticancer potential[88]. Similarly to those describe in the previous section, these studies focused on the ability of the compounds to intercalate between the base pairs of nucleic acids. Thus, synthetized derivatives contained a basic side chain of L-lysine in the positions 9 or 10, and the N-terminal group of L-lysine was linked to the chromophore through an amide bond (Figure 6).
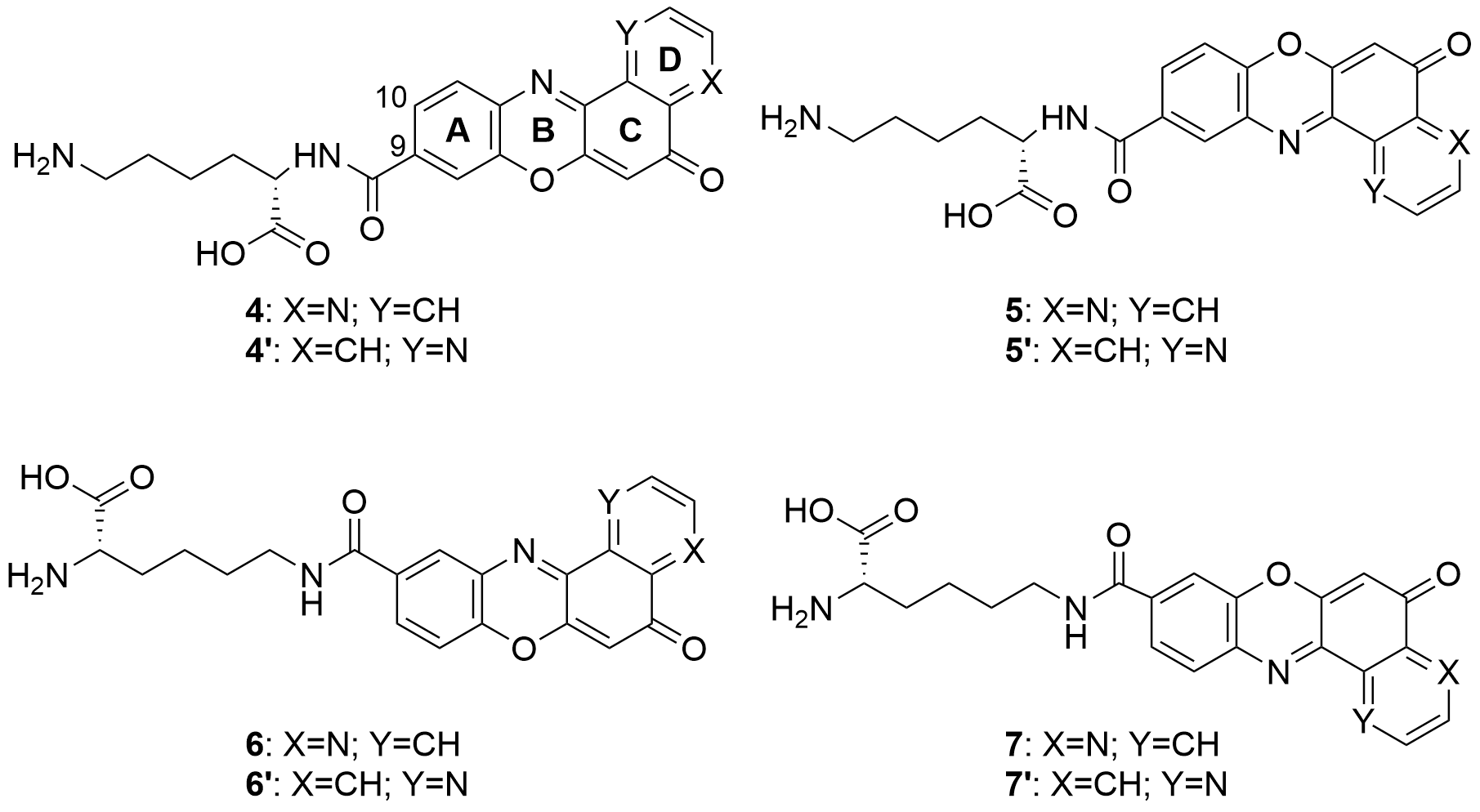
Figure 6. Structure of the pyridophenoxazinone derivatives conjugated to L-lysine.
Products 4-7 and 4’-7’ were tested on cancer cell lines of leukemia (CCRF-CEM, CCRF-SB and MT-4), colon (HT-29), breast cancer (MCF-7), cervical cancer (HeLa), papillary renal cell carcinoma (ACHN), and melanoma (SKMEL-28 and G-361). All the products inhibited the proliferation of a panel of human liquid and solid neoplastic cell lines, the latter being more sensitive to antiproliferative effects (only compound 7 showed similar activity in both states). The IC50 values of the derivatives with the L-lysine side chain attached to C-10 were higher than their corresponding derivative functionalized at C-9. The IC50 values of 4-7 were comparable to that of AMD and even lower than those of Doxo and VP-16 (commercial drugs). It is worth highlighting the lowest values of IC50, all in the range of 0.001-0.007 µM, achieved by 4 and 7 for HT-29, SKMEL-28, MCF-7 and G-361 lines. Authors proved that both compounds are strong DNA intercalators, and possess the capacity to selectively target the topoisomerase Topo IIα.
The difference in activity between the two series of derivatives was related to structure-activity correlations. Thus, the position of the nitrogen atom in D-ring would play an important role in the antiproliferative activity, whereas the position of the L-lysine side chain and the type of amine groups affect the cytotoxic activity both between the two series and within each series.
All the mentioned properties indicate that this new series of pyridophenoxazinones conjugated to L-lysine have a great antitumor therapeutic potential. Products 4 and 7 in particular, provide really interesting opportunities for the development of new DNA-targeting anticancer drugs.
4. Antibacterial and antifungal activities
The emergence of resistant bacteria and its impact on health, together with the inappropriate use of antibiotics, has become a pressing problem for patient safety. Infectious diseases are one of the main causes of global morbidity and mortality, especially in developing countries.
The study of Shimizu et al. (2004)[89] proved that Phx-1, Phx-2 and Phx-3 (Figure 3) can exhibit antibacterial activity. These three molecules were tested on seven non-tuberculous mycobacteria species, being noticeably active against Mycobacterium scrofulaceum attending to minimum inhibitory concentration (MIC) values obtained (1.4-2.8 µg/mL). Phx-3 also inhibited Mycobacterium marinum and Mycobacterium intracellulare, with slightly higher MIC values, whereas Mycobacterium kansasii showed the best inhibition levels for Phx-1 and Phx-2 (Table 1). These results may suggest that the aminophenoxazinones Phx-1, Phx-2 and Phx-3 could be used for the treatment of non-tuberculous mycobacteria infections. The sensitivity to these compounds is related to the characteristics of the cell wall of the strains. Cell wall structure is composed of an innermost electron-dense layer, an electron transparent zone at the intermediate layer and the outermost electron-dense layer (composed of various lipoglycans, free polysaccharides, glycolides and phospholipids)[90].
Table 1. Minimum inhibitory concentration values (µg/mL) obtained by Shimizu et al. (2004) for the compounds Phx-1, Phx-2 and Phx-3 against different species of the genera Mycobacterium
|
Species |
Phx-1 |
Phx-2 |
Phx-3 |
|
M. scrofulaceum |
2.8 |
1.4 |
2.8 |
|
M. marinum |
> 45 |
> 45 |
11.3 |
|
M. intracellulare |
> 45 |
> 45 |
5.6 |
|
M. kansasii |
22.5 |
11.3 |
> 45 |
|
M. tuberculosis, M. fortuitum and M. smegmatis |
> 45 |
> 45 |
> 45 |
In the case of Phx-3, high antituberculosis activity levels has been also reported (MIC = 3.91 µM)[56], as well as promising antiinflammatory and immunoregulatory activities involving in vivo assays in mice[59]. These last conclusions were related with the inhibition of the production of nitric oxide and prostaglandin E2, both achieved with low IC50 values (1.5 and 0.27 µM, respectively). Phx-3 was suggested for the treatment of T cell-mediated autoimmune diseases, and for those chronic-inflammatory diseases induced by bacteria.
4.1 Antibacterial activity on genera Helicobacter
Helicobacter pylori, which affects approximately 50% of the world population, is a causative agent of chronic gastroduodenal ulcer and, possibly, gastric cancer. The triple therapy that comprises amoxicillin, clarithromycin (CAM) and a proton pump inhibitor, has proven to be effective to eradicate this bacterium. However, resistance is a common issue that appears when applied to the control of bacteria like H. pylori. As an example, the recovery rate when this triple therapy treatment is applied in Japan is as low as 20%[91]. This loss of effectiveness caused by resistance phenomena is therefore a serious concern with regard to the eradication of this type of infection[92].
This resistance issue has brought about the development of alternative drugs over the last decade, being Phx-3 one of the drugs of interest for this purpose[93]. Phx-3 was, therefore, tested against standard strains of H. pylori (including 43504 and ATCC 43579) and against three clinical strains (TK 1029, 1047 and 1402). According to the results regarding cell-mediated immunity (CMI), none of the strains, including TK 1029, which has proven to be resistant to the triple therapy, grow in the presence of Phx-3 at concentrations of 1 µg/mL or higher. After 48 h, the bacterial populations were below their detection limit. Phx-3 has also been tested against H. musterae (ATCC 43772), and 0.5 µg/mL CMI value was reported.
Inhibitor compounds of H. pylori have the capacity to implement certain morphological changes[94]. Such changes that take place after the compound has been administered need to be evaluated. Thus, cells in ATCC 43504 strains were subjected to a culture of Phx-3 (6 µg/mL) for 6 hours, after which, severe morphological changes were revealed by scanning electron microscopy, namely the detachment of outer membranes, which might be the mechanism responsible for the rapid bactericidal activity of TG44. In the case of Phx-1 and Phx-2, no activity was found against H. pylori and H. mustelae strains.
Information provided herein would be quite relevant for the development of new drugs for the control of H. pylori based on Phx-3. Some reports associate H. pylori infections to gastric lymphomas and adenocarcinomas (currently known as type 1 carcinogens)[95]. Therefore, the control of this bacterium also contributes to the prevention of certain types of cancers.
4.2 Other antibacterial and antifungal activities
Similarly to Helicobacter genera, the appearance of certain antimicrobial resistance represents a menace to the prevention and treatment of infections caused by bacteria, parasites, viruses and fungi. The diseases associated to these organisms are the third leading cause of death in developed countries[96]. For instance, the virulence of Fusarium spp. (fungi) in plants is conditioned by the resistance generated by the detoxification of certain drugs, like benzoxazolinones BOA and MBOA (plant defense compounds). This process was found to be catalyzed by the fungal γ-lactamase enzyme encoded by FDB1 locus[97]. Therefore, new strategies or compounds that are not affected by this resistance need to be developed.
Phx-3 has shown high levels of toxicity to the species Fusarium verticilloides[98]. Tested at 50 µM dose, it is also active against Bacillus subtilis and Staphylococcus aureus, as well as on the fungi Candida albicans, whereas no activity against Escherichia coli was observed. The hydroxilated derivative at C-6 was active for the two first species, with slightly better results[99]. We would like to recommend to all those who intend to conduct further studies on the activity of Phx-3 against S. aureus the article by Tse et al. (2012)[100]. Later on, a silver(I) complex of Phx-3 (first metal complex reported of this compound to be reported) showed an improvement of the activity against S. aureus, that was attributed to the release of silver ions into the cells after getting dissociated from the complex. Like Phx-3, this complex was inactive against E. coli, which was probably explained by the obstacle posed by the double layer cell wall that characterizes gram-negative species[21].
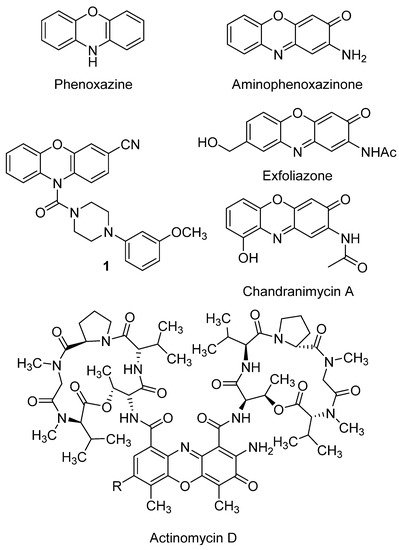
Chandrananimycin A (Figure 2) and B are natural products related to Phx-3, characterized by the presence of an amide function instead of the common exocyclic amine group. Both have shown inhibition activity on Mucor miehei and, in the case of the first, also on B. subtilis[101]. In the same study chandrananimycin C was also evaluated, being active on the same species (increasing almost to the double the inhibition zones) together with S. aureus, C. albicans, Chlorella vulgaris, Chlorella sorokiniana and Scenedesmus suspicatus. The better activity of chandrananimycin C in the concentration tested (20 µM) would be related to its structure, which presents the scaffold of chandrananimycin B with an additional fourth ring (cyclic amine). None of the three compounds were active on E. coli.
Natural product NHP (Figure 4), in addition to the cytotoxicity on cancer lines previously mentioned in section 3.1, has shown antifungal activity against C. albicans (MIC = 64 µg/mL)[60].
Recent studies[102] evaluated the antimicrobial activity of 8 derivatives (8-15) of phenoxazines against the bacteria Listeria ivanovii, E. coli, S. aureus and Klebsiellapneumoniae spp., and against the fungi C. albicans and Aspergillusniger spp. For the synthesis of the compounds, at a first stage, 5 was synthesized, then, the rest of compounds were obtained by arylation (9-12) or amination (13-15) reactions of 8 (Figure 7).
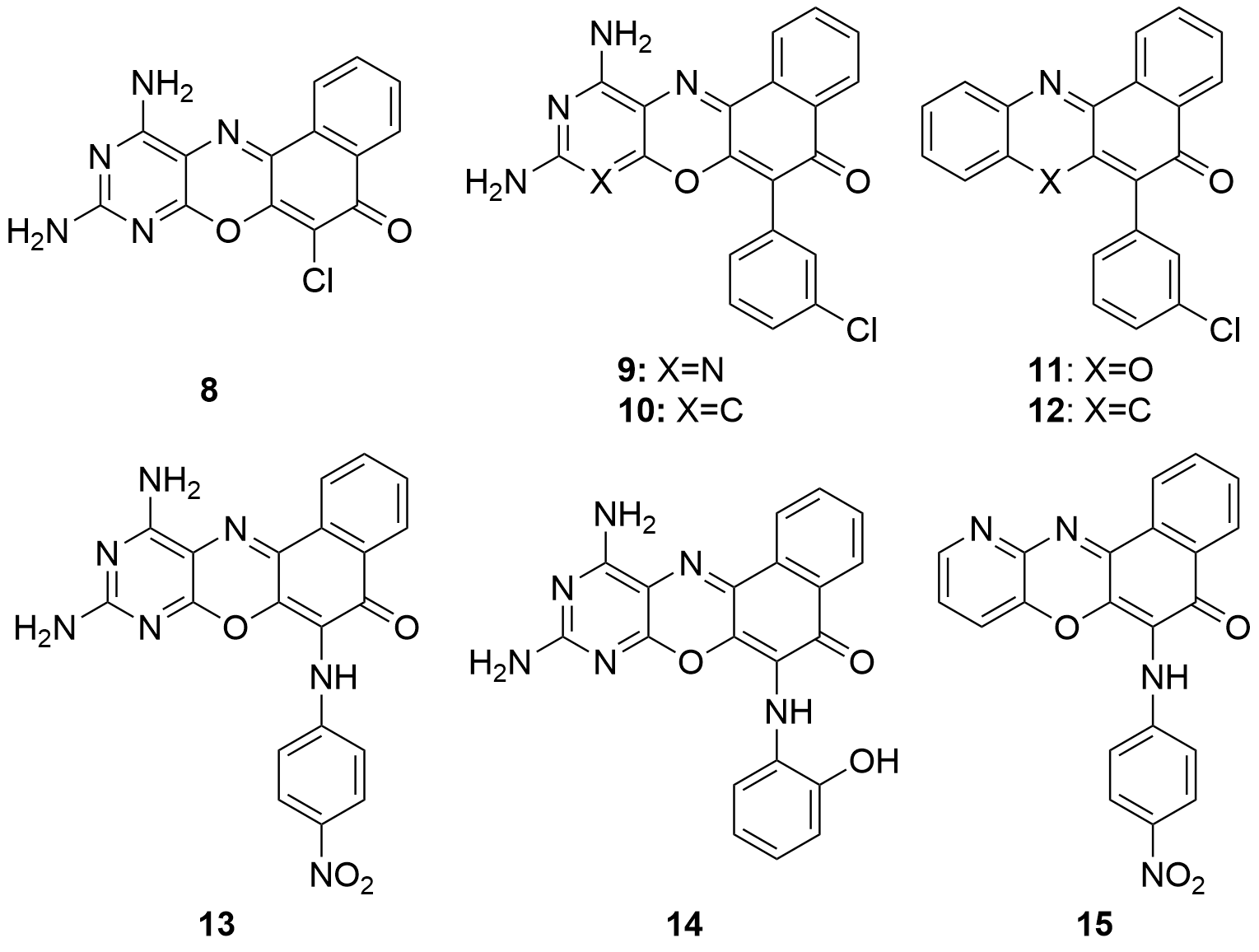
Figure 7. Compound 8 and derivatives obtained by arylation using boronic acids (9-12) or by amination using anilines (13-15).
The agar well diffusion method was employed for the sensitivity study of 8-15. All the compounds showed a considerable activity against L. ivanovii, except compound 12. Only half of the compounds (8, 9, 14 and 15) exhibited activity against E. coli, which was more selective. In the case of S. aureus, K. pneumoniae, C. albicans and A. niger, it should be highlighted that only compound 9 presented some activity against all these species. It should also be noted that compound 8 (scaffold of 9-15) showed sensitivity to all the organisms except S. aureus and L. ivanovii, both of which are gram-positive bacteria. Therefore, it could be concluded, with regard molecular structures, that the compounds obtained either through the arylation or amination of compound 8 causes the inactivation of the molecule on fungi, but exhibits an increased activity when applied to gram-positive bacteria.
The data obtained from the experiments show how most of the compounds are active at low concentrations. The best minimum active concentration observed was for derivatives 11 and 15 on L. ivanonii (14.42 µg/mL), being a significantly lower value than that of the reference drug (21.30 µg/mL). This higher activity than currently commercialized drugs, let us think that new derivatives could be developed to a more efficient control of microorganisms.
Sridhar et al. (2015)[103] reported the evaluation of a family of chlorinated phenoxazine derivatives characterized by the presence of a tertiary amine at the second ring. Each product presents a specific moiety bonded to this nitrogen atom. Four of these products showed to be significantly active on Klebsialla sp., Bacillus sp., Proteus vulgaris, Salmonella sp., Shigella flexneri, S. aureus, Streptococcus pneumoniae and Vibrio cholerae, being remarked compound 16 (Figure 8).
The final study presented in this section will be the finding of new chromogen aminophenoxazinone derivatives with applicability for the detection of Pseudomonas aeruginosa (pathogen gram-negative bacteria) by detecting β-alanyl aminopeptidase activity in clinical samples[104]. This family of compounds is characterized by the presence of a β-alanyl moiety bonded to the first ring of aminophenoxazinones via an amide bond. In the article, it is provided the results of a broad screening test of the compounds on diverse bacterial species of both gram categories.
5. Other activities
5.1 Antiviral activity
In vivo antiviral studies of aminophenoxazinones Phx-1, Phx-2 and Phx-3 in mice allowed to propose the three compounds as potential drugs for preventing the replication of HSV and the aggravation of lesions that these viruses cause. Their activity on herpes simplex virus type-1 (HSV-1) was highlighted, whereas Phx-2 was the only that showed inhibitory effects against HSV-2 (moderately). Another point of interest regarding HSV-2 was the improvement of the survival rates after the treatment with Phx-1, Phx-2 or Phx-3 (without treatment, all the infected did not survived)[105]. Phx-2 also inactivates human cytomegalovirus (HCMV) in vitro, being measured a markedly better value of selectivity index[106]. The inactivity of Phx-3 on H1N1 and H3N2 influenza viruses has been reported[56].
The antiviral potential of the three compounds is also promising for the human T-lymphotropic virus type-1 (HTLV-1), which causes adult T-cell leukemia (ATL). Phx-1, Phx-2 and Phx-3 prevent the cell-to-cell transmission of this virus in model cell lines, as well as the induction of apoptosis in MT-1 cells in doses of 10 μg/mL[107]. The same study suggested that this apoptosis process occurs by post-translational events like the depolarization of mitochondria, as the aminophenozaxizones activated the enzyme caspase-3.
Recently, Phx-3 was computationally studied by docking to evaluate its pharmacokinetic related with the inhibition against MMP9 enzymes, whose overexpression is caused by the infection of hepatitis B virus[108]. As result, Phx-3 would possess key properties under the acceptable range, however, it was not among the most potent compounds evaluated in this study.
5.2 Antiparasitic activity
Malaria is a parasitic disease that causes high fevers, chills, flu-like symptoms and anemia. It is caused by a parasite and is transmitted to humans through the bite of infected Anopheles mosquitoes[109].
Jian-Feng Ge et al. (2010)[110] evaluated the antimalarial activity of various phenoxazin derivatives. The best results were obtained from the derivative functionalized with a 4-aminopyridine group (17, Figure 8). The activity level measured for 18 should also be highlighted. However, this compound presented the disadvantage of its shorter life; probably due to the shorter chains of the tertiary amine.
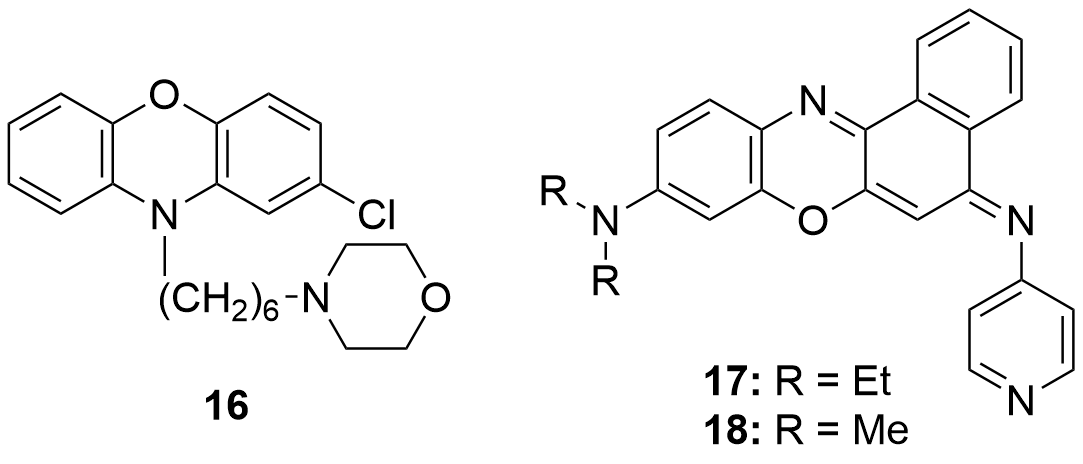
Figure 8. Phenoxazin derivatives with antibacterial (16) and antimalarial (17 and 18) activity.
Compound 17 was considered a suitable candidate for the development of antimalarial drugs. In the first place, it exhibited an IC50 of 7.6 nM it against Plasmodium falciparum as well as a noticeable higher selectivity when compared to the other synthesized derivatives. Moreover, the in vivo experiments that were carried out on mice infected with Plasmodium berghei (the parasitic species that causes malaria in some rodents) achieved the cure after three daily doses for three consecutive days.
Leishmania major is a parasitic species that causes cutaneous leishmaniasis, a tropical disease that generates a large number of adverse effects on human health, mainly affecting the skin and mucous membranes. Human infections occur after the bite of sand fly (Phlebotomus). A number of haloacetamines derivated from N-substituted phenoxazines, as well as other related structures such as phenothiazines, have been studied through in vitro essays[111]. The haloacetamide derivatives proved to be suitable to inhibit L. major. The lowest IC50 values were obtained by the products shown in Figure 9: 7.6 µM (19), 9.5 µM (20) and 9.6 µM (21).
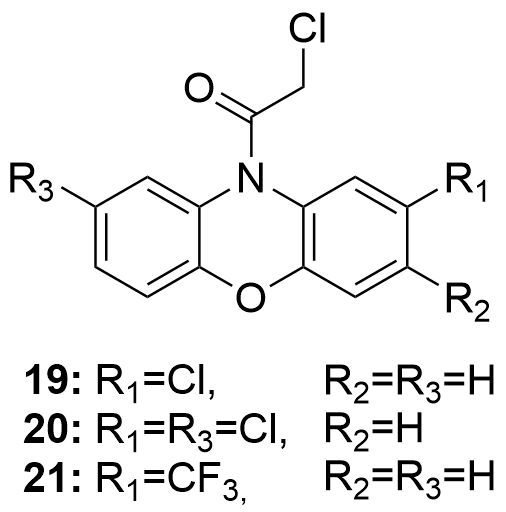
Figure 9. Compounds that have exhibited some interesting potential to treat L. major infections.
6. Conclusions
The compendium of such studies from the literature led us to conclude that aminophenoxazinones possess a number of promising properties like anticarcinogenic, antifungal, antiparasitic, antibacterial or antimicrobial. Aminophenoxazinones are degradation products that present really interesting pharmacological properties, selectivity and minimal toxicity that make them suitable for the treatment of certain current relevant diseases as explained in this review. Phx-1, Phx-2 and Phx-3 are the most studied aminophenoxazinones in the literature, especially Phx-3, whose activities and modes of action have been detailed herein. In the case of the cytotoxicity generated against cancer cell lines, Phx-3 stands out as promising anticancer drug, due to the capacity to decrease the pHi in cancer cells and inhibit the proliferation of cell lines like the related to gastric and colon cancer, glioblastoma or melanoma, among others. The activity of Phx-3 has been compared to that of anticancer drugs like camptothecin, known for its side effects. In this context, in vivo studies have shown that Phx-1 and Phx-3 cause low or null adverse effects, which indicates that they would be less aggressive drugs. Regarding their antibacterial and antimicrobial activities, Phx-1, Phx-2 and Phx-3 have proven to have the desirable characteristics that would allow certain drug resistance issues to be overcome. Some non-tuberculous mycobacteria, H. pylori or F. verticilloides are examples of high sensitive species to these aminophenoxazinones. Moreover, it should be remarked their activity against herpes simplex virus type-1 (HSV-1) and human T-lymphotropic virus type-1 (HTLV-1). Remarkable antiproliferative capacity have been proven for some structural derivatives, such as those conjugated to indole groups 2 and 3 (especially for lung adenocarcinoma), the pyridophenoxazinones 4 and 7 (for melanoma, colon and breast cancer), compounds 8, 9, 11, 15 and 16 (antibacterial or antifungal), 17 (antimalarial) and 19-21 (against Leishmania major).
Therefore, the active compounds remarked herein could be considered for the development or improvement of therapeutic treatments, from the perspective of the activity levels. This conclusion supports the research studies that are currently being conducted on this family of compounds. In authors’ opinion, it is necessary the search and evaluation of new derivatives that allow to define the structural features that provoke the effects on the different types of assays. Even though different in vivo studies had been presented in this review, we consider that the proportion of this type of assays should increase over the next years, which would bring firm conclusions for the future use of aminophenoxazinones and related compounds as drugs.
References
- Sánchez-Moreiras, A.M.; Coba de la Peña, T.; Martínez, A.; González, L.; Pellisier, F.; Reigosa, M.J. Mode of action of the hydroxamic acid BOA and other related compounds. In Allelopathy; Macías, F.A., Galindo, J.C.G., Molinillo, J.M.G., Cutler, H.G., Eds.; CRC Press, Boca Raton, FL., 2004; pp. 239–252.
- Hector R. Bravo; Waldo Lazo; Antialgal and Antifungal Activity of Natural Hydroxamic Acids and Related Compounds. Journal of Agricultural and Food Chemistry 1996, 44, 1569-1571, 10.1021/jf950345e.
- Bettina M. Jensen; Khem Adhikari; Heidi J. Schnoor; Nanna Juel-Berg; Inge S. Fomsgaard; Lars K. Poulsen; Quantitative analysis of absorption, metabolism, and excretion of benzoxazinoids in humans after the consumption of high- and low-benzoxazinoid diets with similar contents of cereal dietary fibres: a crossover study. European Journal of Nutrition 2015, 56, 387-397, 10.1007/s00394-015-1088-6.
- Shuyun Peng; William Scott Chilton; Biosynthesis of dimboa in maize using deuterium oxide as a tracer. Phytochemistry 1994, 37, 167-171, 10.1016/0031-9422(94)85018-6.
- Praveen Kumar; Donald E. Moreland; William Scott Chilton; 2H-1,4-benzoxazin-3(4H)-one, an intermediate in the biosynthesis of cyclic hydroxamic acids in maize. Phytochemistry 1994, 36, 893-898, 10.1016/s0031-9422(00)90458-8.
- Khem Adhikari; Helle N. Lærke; Anne G. Mortensen; Inge S. Fomsgaard; Plasma and Urine Concentrations of Bioactive Dietary Benzoxazinoids and Their Glucuronidated Conjugates in Rats Fed a Rye Bread-Based Diet. Journal of Agricultural and Food Chemistry 2012, 60, 11518-11524, 10.1021/jf301737n.
- Stine Krogh Steffensen; Hans A. Pedersen; Khem B. Adhikari; Bente B. Laursen; Claudia Jensen; Søren Høyer; Michael Borre; Helene H. Pedersen; Mette Borre; David Edwards; et al.Inge Sindbjerg Fomsgaard Benzoxazinoids in Prostate Cancer Patients after a Rye-Intensive Diet: Methods and Initial Results. Journal of Agricultural and Food Chemistry 2016, 64, 8235-8245, 10.1021/acs.jafc.6b03765.
- Praveen Kumar; Ronald W. Gagliardo; William Scott Chilton; Soil transformation of wheat and corn metabolites mboa and DIM2BOA into aminophenoxazinones. Journal of Chemical Ecology 1993, 19, 2453-2461, 10.1007/bf00980682.
- Francisco A. Macias; Alberto Oliveros-Bastidas; David Marín; Diego Castellano; Ana M. Simonet; José M. G. Molinillo; Degradation Studies on Benzoxazinoids. Soil Degradation Dynamics of (2R)-2-O-β-d-Glucopyranosyl-4-hydroxy-(2H)- 1,4-benzoxazin-3(4H)-one (DIBOA-Glc) and Its Degradation Products, Phytotoxic Allelochemicals from Gramineae. Journal of Agricultural and Food Chemistry 2005, 53, 554-561, 10.1021/jf048702l.
- Inge S. Fomsgaard; Anne G. Mortensen; Sandra C.K. Carlsen; Microbial transformation products of benzoxazolinone and benzoxazinone allelochemicals––a review. Chemosphere 2004, 54, 1025-1038, 10.1016/j.chemosphere.2003.09.044.
- Francisco A. Macías; David Marín; Alberto Oliveros-Bastidas; José María González Molinillo; Rediscovering the bioactivity and ecological role of 1,4-benzoxazinones. Natural Product Reports 2009, 26, 478-489, 10.1039/b700682a.
- Francisco A. Macías; Alberto Oliveros-Bastidas; David Marín; Diego Castellano; Ana M. Simonet; José María González Molinillo; Degradation Studies on Benzoxazinoids. Soil Degradation Dynamics of 2,4-Dihydroxy-7-methoxy-(2H)-1,4-benzoxazin-3(4H)-one (DIMBOA) and Its Degradation Products, Phytotoxic Allelochemicals from Gramineae. Journal of Agricultural and Food Chemistry 2004, 52, 6402-6413, 10.1021/jf0488514.
- Sascha Venturelli; Regina G. Belz; Andreas Kämper; Alexander Berger; Kyra Von Horn; André Wegner; Alexander Böcker; Gérald Zabulon; Tobias Langenecker; Oliver Kohlbacher; et al.Fredy BarnecheDetlef WeigelUlrich M. LauerMichael BitzerClaude Becker Plants Release Precursors of Histone Deacetylase Inhibitors to Suppress Growth of Competitors. The Plant Cell 2015, 27, 3175-3189, 10.1105/tpc.15.00585.
- Francisco A. Macías; Alberto Oliveros-Bastidas; David Marín; Nuria Chinchilla; Diego Castellano; José María González Molinillo; Evidence for an Allelopathic Interaction Between Rye and Wild Oats. Journal of Agricultural and Food Chemistry 2014, 62, 9450-9457, 10.1021/jf503840d.
- Nuria Chinchilla; David Marín; Alberto Oliveros-Bastidas; José M. G. Molinillo; Francisco A. Macías; Soil biodegradation of a benzoxazinone analog proposed as a natural products-based herbicide. Plant and Soil 2015, 393, 207-214, 10.1007/s11104-015-2485-6.
- Gierl, A.; Gruen, S.; Genschel, U.; Huettl, R.; Frey, M. Evolution of indole and benzoxazinone biosynthesis in Zea mays. In Recent Advances in Phytochemistry; Elsevier, Ed.; 2004; pp. 69–83.
- Mireia Farres; Marta Villagrasa; Ethel Eljarrat; Damià Barceló; Romà Tauler; Chemometric evaluation of different experimental conditions on wheat (Triticum aestivum L.) development using liquid chromatography mass spectrometry (LC–MS) profiles of benzoxazinone derivatives. Analytica Chimica Acta 2012, 731, 24-31, 10.1016/j.aca.2012.04.017.
- Margot Schulz; Adriano Marocco; Vincenzo Tabaglio; Francisco A. Macias; José María González Molinillo; Benzoxazinoids in Rye Allelopathy - From Discovery to Application in Sustainable Weed Control and Organic Farming. Journal of Chemical Ecology 2013, 39, 154-174, 10.1007/s10886-013-0235-x.
- Tomoda, A., Yamaguchi, T., Sato, K., & Iwata, A. (2004). Antiviral agents containing aminophenoxazines. Jpn. Patent, 2004143101.
- Bitzer, M.; Lauer, U.M.; Venturelli, S.; Armeanu, S. Aminophenoxazinone compounds as antitumor and antiinflammatory agents 2009
- Komala Pandurangan; Shane Gallagher; Grace G. Morgan; Helge Müller-Bunz; Francesca Paradisi; Structure and antibacterial activity of the silver(i) complex of 2-aminophenoxazine-3-one. Metallomics 2010, 2, 530-534, 10.1039/c003515g.
- F. Kehrmann; Ueber Oxydationsproducte vono-Aminophenolen. European Journal of Inorganic Chemistry 1906, 39, 134-138, 10.1002/cber.19060390127.
- Zoltan Szeverényi; Elena R. Milaeva; Lászlò I. Simándi; Kinetics of the oxidation of 2-aminophenol by dioxygen in the presence of tetrakis(3,5-di-t-butyl-4-hydroxyphenyl)-dodecachlorophthalocyaninatocobalt(II). Journal of Molecular Catalysis 1991, 67, 251-258, 10.1016/0304-5102(91)85050-c.
- S. Gabriel; Ueber eine Darstellungsweise primärer Amine aus den entsprechenden Halogenverbindungen. European Journal of Inorganic Chemistry 1887, 20, 2224-2236, 10.1002/cber.18870200227.
- Helge Prinz; Ann-Kathrin Ridder; Kirsten Vogel; Konrad J. Böhm; Igor Ivanov; Jahan B. Ghasemi; Elham Aghaee; Klaus Müller; N-Heterocyclic (4-Phenylpiperazin-1-yl)methanones Derived from Phenoxazine and Phenothiazine as Highly Potent Inhibitors of Tubulin Polymerization. Journal of Medicinal Chemistry 2017, 60, 749-766, 10.1021/acs.jmedchem.6b01591.
- Shun Li; Tianyuan Cheng; Changfeng Yin; Sensen Zhou; Quli Fan; Wei Wu; Xiqun Jiang; Phenothiazine versus Phenoxazine: Structural Effects on the Photophysical Properties of NIR-II AIE Fluorophores. ACS Applied Materials & Interfaces 2020, 12, 43466–43473, 10.1021/acsami.0c12773.
- Wubin Zheng; Jiajia Yang; Yang Shen; Yusi Yao; Guanglei Lv; Shiyou Hao; Chunxia Li; The near-infrared fluorescent probes based on phenoxazine for the rapid detection of hypochlorous acid. Dyes and Pigments 2020, 179, 108404, 10.1016/j.dyepig.2020.108404.
- Luke Farmer; Evan A. Haidasz; Markus Griesser; Derek A. Pratt; Phenoxazine: A Privileged Scaffold for Radical-Trapping Antioxidants. The Journal of Organic Chemistry 2017, 82, 10523-10536, 10.1021/acs.joc.7b02025.
- Efeturi A. Onoabedje; Samuel A. Egu; Mercy A. Ezeokonkwo; Uchechukwu C. Okoro; Highlights of molecular structures and applications of phenothiazine & phenoxazine polycycles. Journal of Molecular Structure 2019, 1175, 956-962, 10.1016/j.molstruc.2018.08.064.
- Carla M.A. Alves; Sarala Naik; Paulo Coutinho; M. Sameiro T. Gonçalves; Novel DNA fluorescence probes based on N-[5-(11-functionalised-undecylamino)-9H-benzo[a]phenoxazin-9-ylidene]propan-1-aminium chlorides: synthesis and photophysical studies. Tetrahedron Letters 2011, 52, 112-116, 10.1016/j.tetlet.2010.10.165.
- Wei-Jin Zhu; Jin-Yun Niu; Dan-Dan He; Ru Sun; Yu-Jie Xu; Jian-Feng Ge; Near-infrared pH probes based on phenoxazinium connecting with nitrophenyl and pyridinyl groups. Dyes and Pigments 2018, 149, 481-490, 10.1016/j.dyepig.2017.09.059.
- Agata Jaszczyszyn; Kazimierz Gąsiorowski; Piotr Świątek; Wiesław Malinka; Katarzyna Cieślik-Boczula; Joanna Petrus; Bogusława Czarnik-Matusewicz; Chemical structure of phenothiazines and their biological activity. Pharmacological Reports 2012, 64, 16-23, 10.1016/s1734-1140(12)70726-0.
- Mei Peng; Yiling Ding; Ling Yu; Yali Deng; Weisi Lai; Yun Hu; Hongwen Zhang; Xianqing Wu; Hong Fan; Hui Ding; et al.Yilin WuGuangshi Tao Tegafur Substitution for 5-Fu in Combination with Actinomycin D to Treat Gestational Trophoblastic Neoplasm. PLOS ONE 2015, 10, e0143531, 10.1371/journal.pone.0143531.
- Layla Mohammad Hadi; Elnaz Yaghini; Alexander J. MacRobert; Marilena Loizidou; Synergy between Photodynamic Therapy and Dactinomycin Chemotherapy in 2D and 3D Ovarian Cancer Cell Cultures. International Journal of Molecular Sciences 2020, 21, 3203, 10.3390/ijms21093203.
- Jonas Kühlborn; Matthias Konhäuser; Jonathan Groß; Peter R. Wich; Till Opatz; Xylochemical Synthesis of Cytotoxic 2-Aminophenoxazinone-Type Natural Products Through Oxidative Cross Coupling. ACS Sustainable Chemistry & Engineering 2019, 7, 4414-4419, 10.1021/acssuschemeng.8b06353.
- Pasceri, R.; Siegel, D.; Ross, D.; Moody, C.J.; Aminophenoxazinones as inhibitors of indoleamine 2,3-dioxygenase (IDO). Synthesis of exfoliazone and chandranimycin A. J. Med. Chem. 2013, 56, 3310–3317, 10.1021/jm4000049z.
- Song Wu; Sarah E Powers; Wenquan Zhu; Yusuf A. Hannun; Substantial contribution of extrinsic risk factors to cancer development. Nature 2015, 529, 43-47, 10.1038/nature16166.
- А. Romaniuk; M. Lyndin; V. Sikora; Y. Lyndina; K. Sikora; Heavy metals effect on breast cancer progression. Journal of Occupational Medicine and Toxicology 2017, 12, 32-32, 10.1186/s12995-017-0178-1.
- Mattiuzzi, C.; Lippi, G.; Current Cancer Epidemiology. J. Epidemiol. Glob. Health 2019, 9, 217–222.
- Japanese Gastric Cancer Association; Japanese gastric cancer treatment guidelines 2018 (5th edition). Gastric Cancer 2020, 24, 1-21, 10.1007/s10120-020-01042-y.
- Yan Zhao; Qinhao Guo; Jiejing Chen; Jun Hu; Shuwei Wang; Yueming Sun; Role of long non-coding RNA HULC in cell proliferation, apoptosis and tumor metastasis of gastric cancer: A clinical and in vitro investigation. Oncology Reports 2013, 31, 358-364, 10.3892/or.2013.2850.
- William Waddingham; Stella A V Nieuwenburg; Sean Carlson; Manuel Rodriguez-Justo; Manon Spaander; Ernst J Kuipers; Marnix Jansen; David G Graham; Matthew Banks; Recent advances in the detection and management of early gastric cancer and its precursors. Frontline Gastroenterology 2020, flgastro-2018, 101089, 10.1136/flgastro-2018-101089.
- Christian Stock; Stine Falsig Pedersen; Roles of pH and the Na + /H + exchanger NHE1 in cancer: From cell biology and animal models to an emerging translational perspective?. Seminars in Cancer Biology 2017, 43, 5-16, 10.1016/j.semcancer.2016.12.001.
- Dolores Pérez-Sala; Dolores Collado-Escobar; Faustino Mollinedo; Intracellular Alkalinization Suppresses Lovastatin-induced Apoptosis in HL-60 Cells through the Inactivation of a pH-dependent Endonuclease. Journal of Biological Chemistry 1995, 270, 6235-6242, 10.1074/jbc.270.11.6235.
- Yue Song; Zhao Wang; Zengping Hao; Lihong Li; Junli Lu; Hongjun Kang; Yanping Lu; Yanqin You; Lijuan Li; Qingyun Chen; et al.Bo Chen Requirement for etoposide in the treatment of pregnancy related hemophagocytic lymphohistiocytosis: a multicenter retrospective study. Orphanet Journal of Rare Diseases 2019, 14, 50, 10.1186/s13023-019-1033-5.
- Kaufmann, S.H.; nduction of Endonucleolytic DNA Cleavage in Human Acute Myelogenous Leukemia Cells by Etoposide, Camptothecin, and Other Cytotoxic Anticancer Drugs: A Cautionary Note. Cancer Res. 1989, 49, 5870–5878.
- Xiao-Fang Che; Shin-Ichi Akiyama; Akio Tomoda; Suppression of the proliferation of cancer cell lines, KB-3-1 and K562 cells preceded by a decrease in intracellular pH caused by phenoxazine derivatives.. Oncology Reports 2008, 19, 1253-1258, 10.3892/or.19.5.1253.
- Hiroshi Mori; Katsuaki Honda; Ryoji Ishida; Tomoyoshi Nohira; Akio Tomoda; Antitumor activity of 2-amino-4,4α-dihydro-4α, 7-dimethyl-3H-phenoxazine-3-one against Meth A tumor transplanted into BALB/c mice. Anti-Cancer Drugs 2000, 11, 653-657, 10.1097/00001813-200009000-00010.
- Naoko Miyano-Kurosaki; Kunihiko Kurosaki; Michiko Hayashi; Hiroshi Takaku; Masaaki Hayafune; Ken Shirato; Teruhiko Kasuga; Takahiko Endo; Akio Tomoda; 2-Aminophenoxazine-3-one Suppresses the Growth of Mouse Malignant Melanoma B16 Cells Transplanted into C57BL/6Cr Slc Mice. Biological and Pharmaceutical Bulletin 2006, 29, 2197-2201, 10.1248/bpb.29.2197.
- Teruhiko Kasuga; Takafumi Tabuchi; Ken Shirato; Kazuhiko Imaizumi; Akio Tomoda; Caspase-independent cell death revealed in human gastric cancer cell lines, MKN45 and KATO III treated with phenoxazine derivatives.. Oncology Reports 2007, 17, 409-415, 10.3892/or.17.2.409.
- Akio Tomoda; Hiroyuki Nagata; Xiao-Fang Che; Keisuke Miyazawa; Masato Konishi; Hideyuki Ubukata; Takafumi Tabuchi; Rapid decrease of intracellular pH associated with inhibition of Na+/H+ exchanger precedes apoptotic events in the MNK45 and MNK74 gastric cancer cell lines treated with 2-aminophenoxazine-3-one. Oncology Reports 2011, 25, 341-346, 10.3892/or.2010.1082.
- Thomas Litman; Stine F. Pedersen; Birte Kramhøft; Torben Skovsgaard; Else K. Hoffmann; pH regulation in sensitive and multidrug resistant Ehrlich ascites tumor cells.. Cellular Physiology and Biochemistry 1998, 8, 138-150, 10.1159/000016277.
- Akio Tomoda; Keisuke Miyazawa; Takafumi Tabuchi; Prevention of Carcinogenesis and Development of Gastric and Colon Cancers by 2-Aminophenoxazine-3-one (Phx-3): Direct and Indirect Anti-Cancer Activity of Phx-3. International Journal of Molecular Sciences 2013, 14, 17573-17583, 10.3390/ijms140917573.
- Mattiuzzi, C.; Lippi, G.; Current Cancer Epidemiology. J. Epidemiol. Glob. Health 2019, 9, 217–222.
- Akio Tomoda; Takeshi Nakachi; Takafumi Tabuchi; Akira Takasaki; Sadao Arai; Keisuke Miyazawa; Anticancer activity of phenoxazines produced by bovine erythrocytes on colon cancer cells. Oncology Reports 2010, 23, 1517-1522, 10.3892/or_00000790.
- Junfeng Wang; Weijun He; Xiaochu Qin; Xiaoyi Wei; Xinpeng Tian; Li Liao; Shengrong Liao; Bin Yang; Zhengchao Tu; Bo Chen; et al.Fazuo WangXiaojiang ZhouYonghong Liu Three new indolyl diketopiperazine metabolites from the antarctic soil-derived fungus Penicillium sp. SCSIO 05705. RSC Advances 2015, 5, 68736-68742, 10.1039/c5ra10828d.
- Xiao-Fang Che; Chun-Lei Zheng; Shin-Ichi Akiyama; Akio Tomoda; 2-Aminophenoxazine-3-one and 2-amino-4,4.ALPHA.-dihydro-4.ALPHA.,7-dimethyl-3H-phenoxazine-3-one cause cellular apoptosis by reducing higher intracellular pH in cancer cells. Proceedings of the Japan Academy, Series B 2011, 87, 199-213, 10.2183/pjab.87.199.
- Takanobu Tabuchi; Xiao-Fang Che; Katsuya Hiraishi; Masakazu Adachi; Kei Miyano; Hideki Sumimoto; Takafumi Tabuchi; Keisuke Miyazawa; Akio Tomoda; Selectively Induced Apoptosis in Human Neutrophils in the Presence of Oxidative Phenoxazines, 2-Amino-4,4α-dihydryo-4α-7H-phenoxazine-3-one and 2-Aminophenoxazine-3-one, Preceded by Decrease of Intracellular pH, Depolarization of the Mitochondria, and Inhibition of Superoxide Generation. Journal of Pharmacological Sciences 2011, 117, 139-148, 10.1254/jphs.11134fp.
- Keizo Kohno; Masaki Miyake; Osamu Sano; Mari Tanaka-Kataoka; Shigeto Yamamoto; Satomi Koya-Miyata; Norie Arai; Mitsukiyo Fujii; Hikaru Watanabe; Shimpei Ushio; et al.Kanso IwakiShigeharu Fukuda Anti-inflammatory and Immunomodulatory Properties of 2-Amino-3H-phenoxazin-3-one. Biological and Pharmaceutical Bulletin 2008, 31, 1938-1945, 10.1248/bpb.31.1938.
- Xiaochun Gao; Yuanyuan Lu; Yingying Xing; Yihua Ma; Jiansheng Lu; Weiwei Bao; Yiming Wang; Tao Xi; A novel anticancer and antifungus phenazine derivative from a marine actinomycete BM-17. Microbiological Research 2012, 167, 616-622, 10.1016/j.micres.2012.02.008.
- Akio Tomoda; Akira Takemura; Xiao-Fang Che; Takafumi Tabuchi; Shota Moriya; Keisuke Miyazawa; Enhancement of cytotoxic and pro-apoptotic effects of 2-aminophenoxazine-3-one on the rat hepatocellular carcinoma cell line dRLh-84, the human hepatocellular carcinoma cell line HepG2, and the rat normal hepatocellular cell line RLN-10 in combination with 2-deoxy-D-glucose. Oncology Reports 2011, 27, 347-355, 10.3892/or.2011.1531.
- Dani S. Bidros; Michael A. Vogelbaum; Novel drug delivery strategies in neuro-oncology. Neurotherapeutics 2009, 6, 539-546, 10.1016/j.nurt.2009.04.004.
- M Azuine; Cancer chemopreventive effect of phenothiazines and related tri-heterocyclic analogues in the 12-O-tetradecanoylphorbol-13-acetate promoted Epstein-Barr virus early antigen activation and the mouse skin two-stage carcinogenesis models. Pharmacological Research 2004, 49, 161-169, 10.1016/j.phrs.2003.07.014.
- Kenia, H.; Shivkumar, B.; Kotnal, R.B.; Ramesha, A.; Devadiga, P.; Simpi, C.C.; Chandrashekar, V.M.; Synthesis and Evaluation of Phenothiazine Derivatives. IOSR J. Pharm. 2020, 10, 54–62.
- Mitsutoshi Nakada; Daisuke Kita; Takuya Watanabe; Yutaka Hayashi; Lei Teng; Ilya V. Pyko; Jun-Ichiro Hamada; Aberrant Signaling Pathways in Glioma. Cancers 2011, 3, 3242-3278, 10.3390/cancers3033242.
- Deepa Soni; James A.J. King; Andrew Henry Kaye; Christopher M. Hovens; Genetics of glioblastoma multiforme: mitogenic signaling and cell cycle pathways converge. Journal of Clinical Neuroscience 2005, 12, 1-5, 10.1016/j.jocn.2004.04.001.
- Marek Los; Subbareddy Maddika; Bettina Erb; Klaus Schulze-Osthoff; Switching Akt: from survival signaling to deadly response. BioEssays 2009, 31, 492-495, 10.1002/bies.200900005.
- Gray Pearson; Fred Robinson; Tara Beers Gibson; Bing-E Xu; Mahesh Karandikar; Kevin Berman; Melanie H. Cobb; Mitogen-Activated Protein (MAP) Kinase Pathways: Regulation and Physiological Functions*. Endocrine Reviews 2001, 22, 153-183, 10.1210/edrv.22.2.0428.
- Davis, R.J. Signal transduction by the JNK group of MAP kinases. In Inflammatory Processes: Molecular Mechanisms and Therapeutic Opportunities; Letts, L.G., Morgan, D.W., Eds.; Birkhäuser Basel: Basel, 2000; pp. 13–21.
- Xiao-Fang Che; Shota Moriya; Chun-Lei Zheng; Akihisa Abe; Akio Tomoda; Keisuke Miyazawa; 2-Aminophenoxazine-3-one-induced apoptosis via generation of reactive oxygen species followed by c-jun N-terminal kinase activation in the human glioblastoma cell line LN229. International Journal of Oncology 2013, 43, 1456-1466, 10.3892/ijo.2013.2088.
- Keisuke Miyazawa; Shota Moriya; Tomohiro Kawaguchi; Xiao-Fang Che; Akio Tomoda; Involvement of endoplasmic reticulum stress-mediated CHOP (GADD153) induction in the cytotoxicity of 2-aminophenoxazine-3-one in cancer cells. International Journal of Oncology 2011, 39, 981-988, 10.3892/ijo.2011.1072.
- Akio Tomoda; Chun-Lei Zheng; Xiao-Fang Che; Shin-Ichi Akiyama; Keisuke Miyazawa; 2-Aminophenoxazine-3-one induces cellular apoptosis by causing rapid intracellular acidification and generating reactive oxygen species in human lung adenocarcinoma cells. International Journal of Oncology 2010, 36, 641-650, 10.3892/ijo_00000540.
- Lindsey Goddard; Lauren Yorozuya; Jane Hirokane; Art of prevention: The importance of melanoma surveillance. International Journal of Women's Dermatology 2020, 6, 257-259, 10.1016/j.ijwd.2020.01.003.
- Jarmila Čelakovská; Josef Bukač; Lenka Čáková; Marie Šimková; Eva Jandová; Epidemiology of Melanoma in the Czech Republic in East Bohemia in the Period 2002–2017 and the Effect of the Annual Sunshine Exposure. Acta Medica (Hradec Kralove, Czech Republic) 2020, 63, 10-17, 10.14712/18059694.2020.10.
- Masaki Miyake; Shigeto Yamamoto; Osamu Sano; Mitsukiyo Fujii; Keizo Kohno; Shimpei Ushio; Kanso Iwaki; Shigeharu Fukuda; Inhibitory Effects of 2-Amino-3H-phenoxazin-3-one on the Melanogenesis of Murine B16 Melanoma Cell Line. Bioscience, Biotechnology, and Biochemistry 2010, 74, 753-758, 10.1271/bbb.90795.
- Wei-Hong Liu; Ming-Gang Li; Yi-Qing Li; Jiang-Yuan Zhao; Zhang-Gui Ding; Pei-Wen Yang; Xiao-Long Cui; Meng-Liang Wen; Cytotoxic metabolites of Streptimonospora salina. Chemistry of Natural Compounds 2008, 44, 503-505, 10.1007/s10600-008-9102-3.
- Tannock, I.F.; Rotin, D.; Acid pH in tumors and its potential for therapeutic exploitation. Cancer Res. 1989, 49, 4373–4384.
- Kayo Machihara; Hidenori Tanaka; Yoshihiro Hayashi; Ichiro Murakami; Takushi Namba; Questiomycin A stimulates sorafenib-induced cell death via suppression of glucose-regulated protein 78. Biochemical and Biophysical Research Communications 2017, 492, 33-40, 10.1016/j.bbrc.2017.08.042.
- Akio Tomoda; Sadao Arai; Ryoji Ishida; Takashi Shimamoto; Kazuma Ohyashiki; An improved method for the rapid preparation of 2-amino-4,4a-dihydro-4a,7-dimethyl-3H-phenoxazine-3-one, a novel antitumor agent.. Bioorganic & Medicinal Chemistry Letters 2001, 11, 1057-1058, 10.1016/s0960-894x(01)00153-6.
- Keisuke Kimura; Yoshihiko Usui; Takaaki Hattori; Naohiko Yamakawa; Hiroshi Goto; Masahiko Usui; Shinya Okada; Ken Shirato; Akio Tomoda; Phenoxazine derivative, 2-amino-4,4α-dihydro-4α,7-dimethyl-3H-phenoxazine-3-one suppresses growth of human retinoblastoma cell line Y79 in vitro and in vivo. Oncology Reports 2008, 19, 3-10, 10.3892/or.19.1.3.
- Shimamoto, T.; Tomoda, A.; Ishida, R.; Ohyashiki, K.; Antitumor effects of a novel phenoxazine derivative on human leukemia cell lines through activation of caspase-3 and telomerase. Clin. Cancer Res. 2001, 7, 704–708.
- Ken Shirato; Kazuhiko Imaizumi; Akihisa Abe; Akio Tomoda; Phenoxazine Derivatives 2-Amino-4,4.ALPHA.-dihydro-4.ALPHA.-phenoxazine-3-one and 2-Aminophenoxazine-3-one-Induced Apoptosis through a Caspase-Independent Mechanism in Human Neuroblastoma Cell Line NB-1 Cells. Biological and Pharmaceutical Bulletin 2007, 30, 331-336, 10.1248/bpb.30.331.
- Francisco J. R. Mejías; Alexandra G. Durán; Jesús G. Zorrilla; Rosa M. Varela; José M. G. Molinillo; Manuel M. Valdivia; Francisco A. Macías; Acyl Derivatives of Eudesmanolides To Boost their Bioactivity: An Explanation of Behavior in the Cell Membrane Using a Molecular Dynamics Approach. ChemMedChem 2020, 16, 1297-1307, 10.1002/cmdc.202000783.
- Mehak Chauhan; Anjali Saxena; Biswajit Saha; An insight in anti-malarial potential of indole scaffold: A review. European Journal of Medicinal Chemistry 2021, 218, 113400, 10.1016/j.ejmech.2021.113400.
- Navriti Chadha; Om Silakari; Indoles as therapeutics of interest in medicinal chemistry: Bird's eye view. European Journal of Medicinal Chemistry 2017, 134, 159-184, 10.1016/j.ejmech.2017.04.003.
- Kamaldeep Paul; Shweta Bindal; Vijay Luxami; Synthesis of new conjugated coumarin–benzimidazole hybrids and their anticancer activity. Bioorganic & Medicinal Chemistry Letters 2013, 23, 3667-3672, 10.1016/j.bmcl.2012.12.071.
- Saiprasad N. Nunewar; Naveen Kotla; Jaya Lakshmi Uppu; Apoorva Dixit; Venkatesh Pooladanda; Chandraiah Godugu; Neelima D. Tangellamudi; Synthesis of 1-(Indol-2-yl)-phenoxazine hybrids from quinacetophenone precursors and their biological evaluation as DNA intercalating agents. Journal of Molecular Structure 2020, 1217, 128311, 10.1016/j.molstruc.2020.128311.
- Silvana Pedatella; Carmen Cerchia; Michele Manfra; Anna Cioce; Adele Bolognese; Antonio Lavecchia; Antitumor agents 7. Synthesis, antiproliferative activity and molecular modeling of new l-lysine-conjugated pyridophenoxazinones as potent DNA-binding ligands and topoisomerase IIα inhibitors. European Journal of Medicinal Chemistry 2020, 187, 111960, 10.1016/j.ejmech.2019.111960.
- Shigetaka Shimizu; Mamoru Suzuki; Akio Tomoda; Sadao Arai; Haruhiko Taguchi; Tomoko Hanawa; Shigeru Kamiya; Phenoxazine Compounds Produced by the Reactions with Bovine Hemoglobin Show Antimicrobial Activity Against Non-tuberculosis Mycobacteria. The Tohoku Journal of Experimental Medicine 2004, 203, 47-52, 10.1620/tjem.203.47.
- Vissa, V.D.; Brennan, P.J.; The genome of Mycobacterium leprae: a minimal myocobacte¬rial gene set. Genome Biol. 2001, 2, reviews1023.1–1023.8, 10.1186/gb-2001-2-8-reviews1023.
- Masahiro Asaka; Toshiro Sugiyama; Mototsugu Kato; Kiichi Satoh; Hajime Kuwayama; Yoshihiro Fukuda; Toshio Fujioka; Tadayoshi Takemoto; Ken Kimura; Takashi Shimoyama; et al.Kihachiro ShimizuShinichi Kobayashi A Multicenter, Double-Blind Study on Triple Therapy with Lansoprazole, Amoxicillin and Clarithromycin for Eradication of Helicobacter pylori in Japanese Peptic Ulcer Patients. Helicobacter 2001, 6, 254-262, 10.1046/j.1523-5378.2001.00037.x.
- Soni, M.; Sharma, P.; Bhadauria, R.S.; Choudhary, M.L.; A Review on Antibacterial Resistance. Pharm. Chem. J. 2020, 7, 68–71.
- Tomoko Hanawa; Takako Osaki; Taki Manzoku; Minoru Fukuda; Hayato Kawakami; Akio Tomoda; Shigeru Kamiya; In Vitro Antibacterial Activity of Phx-3 against Helicobacter pylori. Biological and Pharmaceutical Bulletin 2010, 33, 188-191, 10.1248/bpb.33.188.
- Osamu Kamoda; Kinsei Anzai; Jun-Ichi Mizoguchi; Masatoshi Shiojiri; Toshiharu Yanagi; Takeshi Nishino; Shigeru Kamiya; In Vitro Activity of a Novel Antimicrobial Agent, TG44, for Treatment of Helicobacter pylori Infection. Antimicrobial Agents and Chemotherapy 2006, 50, 3062-3069, 10.1128/aac.00036-06.
- Antonia R. Sepulveda; Helicobacter, Inflammation, and Gastric Cancer. Current Pathobiology Reports 2013, 1, 9-18, 10.1007/s40139-013-0009-8.
- Mohamed A. Ibrahim; Siva S. Panda; Alexander A. Oliferenko; Polina V. Oliferenko; Adel S. Girgis; Mohamed Elagawany; F. Zehra Küçükbay; Chandramukhi S. Panda; Girinath G. Pillai; Ahmed Samir; et al.Kaido TämmC. Dennis HallAlan R. Katritzky Macrocyclic peptidomimetics with antimicrobial activity: synthesis, bioassay, and molecular modeling studies. Organic & Biomolecular Chemistry 2015, 13, 9492-9503, 10.1039/c5ob01400j.
- Kettle, A.J.; Carere, J.; Batley, J.; Benfield, A.H.; Manners, J.M.; Kazan, K.; Gardiner, D.M.; A γ-lactamase from cereal infecting Fusarium spp. catalyses the first step in the degrdation of the benzoxazolinone class of phytoalexins. Fungal Genet. Biol. 2015, 83, 1-9.
- Charles W. Bacon; Dorothy M. Hinton; Anthony Glenn; Francisco A. Macías; David Marin; Interactions of Bacillus mojavensis and Fusarium verticillioides with a Benzoxazolinone (BOA) and its Transformation Product, APO. Journal of Chemical Ecology 2007, 33, 1885-1897, 10.1007/s10886-007-9347-5.
- Jens Bitzer; Thomas Grosse; Linzhu Wang; Siegmund Lang; Winfried Beil; Axel Zeeck; New Aminophenoxazinones from a Marine Halomonas sp.: Fermentation, Structure Elucidation, and Biological Activity. The Journal of Antibiotics 2006, 59, 86-92, 10.1038/ja.2006.12.
- Herman Tse; Elaine Chan; Ching-Wan Lam; Ka-Fai Leung; Pat Chow; Kim-Chung Lee; Kong-Hung Sze; Stanley K. K. Cheung; Man-Kit Tse; Pak Leung Ho; et al.Sze-Pui LeungSusanna K. P. LauPatrick C. Y. WooKwok-Yung Yuen Production of 2-Aminophenoxazin-3-one by Staphylococcus aureus Causes False-Positive Results in -Galactosidase Assays. Journal of Clinical Microbiology 2012, 50, 3780-3782, 10.1128/jcm.02299-12.
- Rajendra P. Maskey; Fuchao C. Li; Song Qin; Heinz H. Fiebig; Hartmut Laatsch; Chandrananimycins A-C: Production of Novel Anticancer Antibiotics from a Marine Actinomadura sp. Isolate M048 by Variation of Medium Composition and Growth Conditions. The Journal of Antibiotics 2003, 56, 622-629, 10.7164/antibiotics.56.622.
- Simon Sani Ocholi; Efeturi Abraham Onoabedje; Samuel Attah Egu; Synthesis and Antimicrobial Studies of 6-Aryl and 6-Anilino Benzo[a]phenoxazinones. European Journal of Advanced Chemistry Research 2020, 1, 1-6, 10.24018/ejchem.2020.1.2.4.
- Sridhar, B.T.; Girish, K.; Channu, B.C.; Thimmaiah, K.N.; Kumara, M.N.; Antibacterial activity of phenoxazine derivatives. J. Chem. Pharm. Res. 2015, 7, 1074–1079.
- Alexandre F. Bedernjak; Andrey V. Zaytsev; Michèle Babolat; Marie Cellier; Arthur L. James; Sylvain Orenga; John D. Perry; Paul W. Groundwater; Rosaleen J. Anderson; Synthesis and Evaluation of Novel 7- and 8-Aminophenoxazinones for the Detection of β-Alanine Aminopeptidase Activity and the Reliable Identification of Pseudomonas aeruginosa in Clinical Samples. Journal of Medicinal Chemistry 2016, 59, 4476-4487, 10.1021/acs.jmedchem.5b01591.
- Kyoko Hayashi; Toshimitsu Hayashi; Keisuke Miyazawa; Akio Tomoda; Phenoxazine derivatives suppress the infections caused by herpes simplex virus type-1 and herpes simplex virus type-2 intravaginally inoculated into mice.. Journal of Pharmacological Sciences 2010, 114, 85-91, 10.1254/jphs.10027fp.
- Kyoko Hayashi; Toshimitsu Hayashi; Akio Tomoda; Phenoxazine Derivatives Inactivate Human Cytomegalovirus, Herpes Simplex Virus-1, and Herpes Simplex Virus-2 In Vitro. Journal of Pharmacological Sciences 2008, 106, 369-375, 10.1254/jphs.fp0071679.
- Naoko Miyano-Kurosaki; Kou Ikegami; Kunihiko Kurosaki; Takahiko Endo; Hoshimi Aoyagi; Mari Hanami; Jun Yasumoto; Akio Tomoda; Anticancer Effects of Phenoxazine Derivatives Revealed by Inhibition of Cell Growth and Viability, Disregulation of Cell Cycle, and Apoptosis Induction in HTLV-1–Positive Leukemia Cells. Journal of Pharmacological Sciences 2009, 110, 87-97, 10.1254/jphs.08347fp.
- Divya, M.; Aparna, C.; Mayank, R.; Singh, M.P.; In-silico insights to identify the bioactive compounds of edible mushrooms as potential MMP9 inhibitor for Hepatitis-B. Res. J. Biotechnol. 2021, 16, 116–126.
- Ansong, D.; Seydel, K.B.; Taylor, T.E. Malaria. In Hunter’s Tropical Medicine and Infectious Disease; Ryan, E.T., Hill, D.R., Solomon, T., Aronson, N.E., Endy, T.P., Eds.; Elsevier: Philadelphia, PA, 2020; pp. 734–754.
- Jian-Feng Ge; Chika Arai; Mei Yang; Abu Bakar; Jun Lu; Nasser S. M. Ismail; Sergio Wittlin; Marcel Kaiser; Reto Brun; Susan A. Charman; et al.Tien NguyenJulia MorizziIsamu ItohMasataka Ihara Discovery of Novel Benzo[a]phenoxazine SSJ-183 as a Drug Candidate for Malaria. ACS Medicinal Chemistry Letters 2010, 1, 360-364, 10.1021/ml100120a.
- Ana Marcu; Uta Schurigt; Klaus Müller; Heidrun Moll; R. Luise Krauth-Siegel; Helge Prinz; Inhibitory effect of phenothiazine- and phenoxazine-derived chloroacetamides on Leishmania major growth and Trypanosoma brucei trypanothione reductase. European Journal of Medicinal Chemistry 2016, 108, 436-443, 10.1016/j.ejmech.2015.11.023.