Severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2) is a novel coronavirus that first appeared in Wuhan, Hubei Province, China in December 2019, connected to a seafood market
[1][2][1,2]. Seven coronaviruses have been reported to infect humans; four of them, human CoV-NL63
[3], HCoV-OC43
[4][5][6][4,5,6], HCoV-229E
[7][8][7,8], and HCoV-HKU
[9][10][9,10], cause mild and seasonal respiratory tract disease, whereas SARS-CoV
[11][12][13][14][15][11,12,13,14,15], MERS-CoV
[16][17][18][19][16,17,18,19], and SARS-CoV-2 can cause severe symptoms. In particular, SARS- CoV-2 is suited to human-to-human transmission and spreads rapidly to other locations, causing lung injury, multiorgan failure, and death
[20][21][20,21]. As of this date, the number of confirmed cases is still increasing, as is the number of deaths
[22][23][22,23]. Therefore, an understanding of the SARS-CoV-2 host and pathogen biology is important to offer valuable insights into the diagnosis and treatment of the disease including the development of new therapies
[24][25][24,25]. Here, we review the basic biology of SARS-CoV-2 including the origin, pathophysiology, and diagnosis methods.
1.1. Nomenclature of SARS-CoV-2
Currently, almost a million sequences of the SAR-CoV-2 genome are publicly available via the Global Initiative on Sharing All Influenza Data (GISAID) and GenBank
[26][27][26,27]. Based on these genome sequences, the phylogenetic classification of SARS-CoV-2 was performed and the nomenclature of GISAID, Phylogenetic Assignment of Named Global Outbreak LINeages (PANGO lineage), and Nextstrain are widely used in scientific and clinical communities
[26][28][29][26,28,29] The major lineages of each nomenclature are summarized in .
Table 1. Nomenclature of SARS-CoV-2.
| GISAID Clades |
PANGO Lineage |
Nextstrain Clades |
Notable Variants |
| S |
A |
19B |
A.23.1 |
| L |
B |
19A |
Wuhan-Hu-1 |
| V |
|
|
|
| G |
B.1 |
20A |
B.1.525, B.1.627 |
| GH |
B.1 |
20C |
B.1.427, B.1.429, B.1.526 |
| |
B.1.2 |
20G |
|
| |
B.1.596 |
|
|
| |
B.1.351 |
20H/501.Y.V2 |
B.1.351 |
| GR |
B.1.1.1 |
20B |
|
| |
P.3 |
|
P.3 |
| |
C |
20D |
|
| |
D.2 |
20F |
|
| |
P.1 |
20J/501.Y.V3 |
P.1 |
| GV |
B.1.177 |
20E (EU1) |
B.1.177 |
| GRY |
B.1.1.7 |
20I/501.Y.V1 |
B.1.1.7 |
GISAID introduced the nomenclature system of SARS-CoV-2 based on marker mutations and named the clade with actual letters of marker mutations
[30]. For example, the clade G has a characteristic mutation in the spike protein gene, D614G. In the nomenclature of GISAID, the initial strains of SARS-CoV-2 were grouped as S and L clades and the current strains of SARS-CoV-2 were classified as eight major clades (S, L, V, G, GH, GR, GV, and GRY)
[30][31][30,31]. The L clade contains the reference strain WIV-04 and was the dominant lineage in early 2020. The L clade later diverged into clades V and G, and clade G diverged into clades GH, GR, GV, and GRY.
The PANGO nomenclature systems focused on the active virus lineage
[29]. This nomenclature is dynamic and the lineages of the PANGO nomenclature are marked as three statuses: active, unobserved, or inactive. The lineages documented within a month are marked as active. The lineages documented within three months are marked as unobserved, and the lineages that were not documented for more than three months are regarded as inactive. The lineages of PANGO nomenclature are named with a letter and numerical values. The initial lineages are denoted as lineages A and B. Although clade B includes the first genome sequenced strain, the phylogenetic analysis suggested that the most recent common ancestor of SARS-CoV-2 was close to early lineage A
[29]. The descendent lineages from initial lineages were assigned with numerical labels. The descendent lineages can be designated with the phylogenetic evidence that the descendent emerged from parental lineages and the descendants showed significant transmission to geographically distinct populations. The designated descendent lineages can also be parental of new emerging lineages and these new lineages have been labeled as parental lineages with additional numerical values. For example, a new emerging lineage from lineage B1 can be labeled as B1.1. The lineages can have a maximum of three sublevels and newly designated lineages emerging from a lineage with three sublevels will be labeled with new alphabetical letters. For example, the parental lineage of lineage C.1 is the lineage B.1.1.1.
The clades of Nextstrain nomenclature were initially named according to year–letter combinations
[32]. Major clades were designated as the clade reached more than 20% of global frequency for more than two months. Based on this criteria, the initial clades were designated as 19A, 19B, 20A, 20B, and 20C. However, due to the global travel restriction, no more clades were designated according to the criteria. For this reason, Nextstrain updated their major clade designation criteria with regional frequency (>30%) and recognized variants of concern. Currently, 12 major clades (19A, 19B, 20A, 20B, 20C, 20D, 20E, 20F, 20G, 20H/501Y.V2, 20I/501Y.V1, 20J/501Y.V3) are designated in the nomenclature of Nextstrain.
1.2. Notable Variants of SARS-CoV-2
The first strain of SARS-CoV-2 was discovered in Wuhan, China and designated as Wuhan-Hu-1 or WIV-04
[1][33][1,33]. The comparison of whole genome sequences showed that the strain was closest to the SARS-like coronavirus RATG13 found in bats (
Rhinolophus affinis) in China
[2]. The overall genomic sequence similarity of RATG13 to SARS-CoV-2 was 96.1%. However, the spike protein gene of RATG13 lacked the furin cleavage site that is essential for the cell entry of SARS-CoV-2, indicating that RATG13 was not the immediate ancestor of SARS-CoV-2
[34][35][34,35]. After discovery of the first SARS-CoV-2 strains, SARS-CoV-2 like viruses were found in pangolins and bats
[36][37][38][36,37,38]. The genome sequences of pangolin-derived CoVs also showed high similarity to those of SARS-CoV-2, but the furin cleavage site was missing in the spike gene sequences of pangolin-derived CoVs
[35][37][38][35,37,38]. A bat-derived CoV, RmYN02, was identified and the genome of the virus showed high similarity to that of SARS-CoV-2
[36]. Although the sequence similarity of RmYN02 was slightly lower than those of RATG13 and pangolin-derived CoVs, the furin cleavage site was inserted, indicating that the addition of the cleavage site can occur naturally
[35][36][35,36].
The D614G variants had a change in spike gene and replaced the initial strains of SARS-CoV-2
[39]. The studies on the variant D614G showed that the infectivity of the variant was increased without increased severity
[39][40][41][39,40,41]. The engineered variants containing the D614G substitution showed more efficient infection in human cells and animal models without altering antibody neutralization and pathogenicity
[41]. A population genetic analysis of COVID-19 also showed that the transmissibility of the variant was increased but there was no sign of increased mortality or clinical severity of the variants
[40].
As new variants with increased pathogenicity, reduced neutralization, and/or increased transmissibility emerged, the U.S. Centers for Disease Control and Prevention (CDC) and Public Health England (PHE) classified some notable variants according to the attributes of the variants
[42][43][42,43]. The CDC classified the variants according to the evidence and significance of the variants into Variant of Interest, Variant of Concern, and Variant of High Consequences. PHE classified variants as Variant Under Investigation (VUI) and Variants Of Concern (VOC). When the variants are considered to have concerning characteristics, they are designated as VUI. After a risk assessment of VUI is conducted, they can be re-designated as VOC. These notable variants are summarized in .
Table 2. Notable variants of SARS-CoV-2.
| PANGO Lineage |
CDC Designation |
PHE Designation |
First Detected |
Spike Protein Substitutions |
| B.1.1.7 |
VOC |
VOC-20DEC-01, VOC-21FEB-02 * |
United Kingdom |
69del, 70del, 144del, (E484K), (S494P), N501Y, A570D, D614G, P681H, T716I, S982A, D1118H (K1191N) |
| B.1.351 |
VOC |
VOC-20DEC-02 |
South Africa |
D80A, D215G, 241del, 242del, 243del, K417N, E484K, N501Y, D614G, A701V |
| P.2 |
VOI |
VUI-21JAN-01 |
Brazil |
E484K, (F565L), D614G, V1176F |
| P.1 |
VOC |
VOC-21JAN-02 |
Brazil |
L18F, T20N, P26S, D138Y, R190S, K417T, E484K, N501Y, D614G, H655Y, T1027I |
| A.23.1 |
- |
VUI-21FEB-01 * |
Uganda |
F157L, V367F, (E484K), Q613H, P681R |
| B.1.525 |
VOI |
VUI-21FEB-03 |
United Kingdom |
A67V, 69del, 70del, 144del, E484K, D614G, Q677H, F888L |
| B.1.1.318 |
- |
VUI-21FEB-04 |
United Kingdom |
D614G, D796H, E484K, P681H, T95I, 144del |
| P.3 |
- |
VUI-21MAR-02 |
Philippines |
E484K, N501Y, P681H |
| B.1.617 |
VOI |
VUI-21APR-01 |
India |
L452R, E484Q, D614G |
| B.1.617.2 |
VOI |
VOC-21APR-02 |
India |
T19R, (G142D), 156del, 157del, R158G, L452R, T478K, D614G, P681R, D950N |
| B.1.617.3 |
VOI |
VUI-21APR-03 |
India |
T19R, G142D, L452R, E484Q, D614G, P681R, D950N |
| AV.1 |
- |
VUI-21MAY-01 |
United Kingdom |
D80G, T95I, G142D, 144del, N439K, E484K, D614G, P681H, I1130V, D1139H |
| B.1.617.1 |
VOI |
- |
India |
(T95I), G142D, E154K, L452R, E484Q, D614G, P681R, Q1071H |
| B.1.526 |
VOI |
- |
United States |
(L5F), T95I, D253G, (S477N), (E484K), D614G, (A701V) |
| B.1.526.1 |
VOI |
- |
United States |
D80G, 144del, F157S, L452R, D614G, (T791I), (T859N), D950H |
| B.1.427 |
VOC |
- |
United States |
L452R, D614G |
| B.1.429 |
VOC |
- |
United States |
S13I, W152C, L452R, D614G |
VOC-20DEC-01, also known as 20I/501Y.V1 or B.1.1.7, was first discovered in the United Kingdom in December 2020
[42] and is defined by 13 mutations
[42]. Recent studies have estimated that the transmissibility of VOC-20DEC-01 is increased by 43–90% and a similar transmission increase was observed globally
[44]. VOC-20DEC-01 was also detected in domestic cats and dogs, raising concern over human-to-animal transmission or vice versa
[45]. Previously reported animal infections were asymptomatic to mild symptomatic, but VOC-20DEC-01 infection in animals showed relatively severe symptoms such as myocarditis
[45]. In February 2021, the variants with a spike gene E484K mutation were reported and designated as VOC-202102/02
[42]. Another variant with N501Y mutation is VOC-20DEC-02 (20H/501Y.V2, or B.1.351), which was first discovered in South Africa. VOC-20DEC-02 is defined by 17 mutations including the E484K mutation, K417N mutation, and two deletions. The variant also showed increased transmissibility (approximately 50%) compared to previous variants
[44]. The third variant with N501Y is VOC-21JAN-02 (P.1 or 20J/501Y.V3), discovered in Brazil
[42][46][42,46]. The genome of VOC-21JAN-02 is defined with 17 non-synonymous mutations, four synonymous mutations, three deletions, and four insertions
[42]. VOC-202101/02 almost fully replaced its parental variant within two months, indicating increased transmissibility of VOC-21JAN-02
[47][48][47,48]. Molecular clock analysis showed that the variants emerged in mid-November 2020 at which time hospitalizations rapidly increased
[49].
The characteristic mutations (N501Y, E484K, and K417N) of the variants with N501Y are mutations in binding sites to viral receptor ACE2 and were already a concern prior to the discovery of these variants
[50][51][52][53][50,51,52,53]. The studies on these variants showed that they had impacts on neutralization by immunity
[54][55][56][57][58][59][60][54,55,56,57,58,59,60]. However, recent research showed that the residual immunity still provided protection, although variants reduced the efficacy of the vaccine
[61].
There were also emerging variants without N501Y, E484K, and/or K417N. The characteristic mutations of A.23.1 are F157L, V367F, Q613H, and P681R
[62]. A.23.1 with E484K was designated as VUI-21FEB-01 in the United Kingdom. These strains were first identified in Uganda and are spreading. One of the characteristic mutations, Q613H, is regarded as functionally equivalent to the D614G mutation of ‘G’ clade strains. B.1.427 and B.1.429 were first discovered and designated as Variants of Concern in the United States
[43]. The characteristic mutations of both lineages are L452R and D614G; these variants showed increased transmissibility and reduced neutralization by convalescent and post-vaccination sera
[63].
B.1.617 was the emerging lineage in India and also designated as VUI-21APR-01 in the United Kingdom
[42]. B.1.167 has two characteristic mutations of different lineages: L452R and E484Q
[64]. The variants were neutralized with convalescent sera of COVID-19 patients and vaccine of BBV152, although the efficacy was low
[64].
2. PCR-Based SARS-CoV-2 Detection
2.1. Reverse Transcription Quantitative PCR (RT-qPCR) Method
Detection of the SARS-CoV-2 viral genome, consisting of single-stranded RNA, is effectively done by reverse transcription quantitative polymerase chain reaction (RT-qPCR), which is the gold standard technique widely used in molecular diagnostics
[65][66][65,66]. There are several practical considerations when performing diagnostic assays using RT-qPCR.
(1) Sample quality: RT-qPCR tests are presently being used for the identification of SARS-CoV-2 in clinical specimens such as upper respiratory tract specimens (saliva, oropharyngeal swab-OPS, nasopharyngeal swab-NPS, nasal swabs), lower respiratory specimens (sputum, bronchoalveolar lavage-BAL, endotracheal aspirate-ET, fibrobronchoscope brush biopsy-FBB), blood (serum, plasma), urine, feces, rectal/anal swabs, stool, and corneal secretion
[67][68][67,68]. To check the sample quality of clinical specimens from different origins, an RNA isolation procedure is required to obtain purified high-quality RNA from the samples, which then needs to be analyzed using chip-based capillary electrophoresis (such as the Agilent Bioanalyzer system), electrophoretic separation on a high-resolution agarose gel, and spectrophotometry
[69].
(2) Reference curve: Data processing can critically affect the analysis of RT-qPCR results
[70]. PCR data processing is based on standard curves or on PCR efficiency assessments
[70]. Standard curves are used to assess RT-qPCR efficiency without theoretical and practical problems
[70]. The estimation of RT-qPCR efficiency using standard curves usually involves the serial dilution of a concentrated stock solution, after which standard samples are analyzed through RT-qPCR by measuring the quantification cycle (Cq) using standard procedures
[70]. The most widely used Cq value is the threshold cycle (Ct), the cycle at which the expression of a target gene first exceeds a calculated fluorescence threshold level
[71]. For example, to detect low amounts of SARS-CoV-2 RNA, a series of diluted RNA templates are used to determine the Ct value, which can provide a standard curve for evaluating the reaction efficiency
[72]. However, the Ct value itself cannot be directly explained as viral load without a standard curve using reference materials
[73]. When interpreting the results of SARS-CoV-2 RT-qPCR, the validity of the standard curve should be proved using reference materials with accurate viral copy numbers to interpret Ct values as viral loads
[73].
(3) Viral load: The success of virus isolation depends on the viral load
[74]. Viral loads in sputum samples and throat swabs are high when obtained within seven days after initial symptoms are observed, ranging from 10
4 to 10
7 copies per mL. This pattern is broken as low quantity of virus are obtained from samples taken after day 8
[75]. In general, sputum samples show higher viral loads than throat swab samples, whereas low viral RNA is detected in urine or stool samples
[75]. The two main factors that influence the quantitative measurement of viral roads are Cq values that are repeatable with acceptable uncertainty and a reliable means of converting from the Ct value to viral load
[76][77][78][76,77,78]. For molecular diagnostic assays, a limit of detection (LoD) and a limit of quantification (LoQ) are also considered the lowest concentrations of target RNA that can be detected by RT-qPCR
[79].
(4) Sampling methods: RT-qPCR tests for SARS-CoV-2 have shown a high variation of false-negative rates (FNR) and false-positive rates (FPR)
[80][81][80,81]. Numerous methods have been developed with the goal of improving the sensitivity, safety, and rapidity of COVID-19 tests by RT-qPCR. For example, one group tested the efficiency and sensitivity of SARS-CoV-2 detection of clinical specimens collected directly in nucleic acid stabilization and lysis buffer (NSLB), a mixture of lysis buffer and RNA preservative, instead of a viral transport medium (VTM), thus inactivating the virus immediately after sampling
[82].
(5) Sample source: To improve the expandability of SARS-CoV-2 testing, several sampling approaches have been developed including nasal, pooled nasal, and throat (oropharyngeal) swabs as well as saliva. Different clinical sampling methods affect the diagnostic performance of SARS-CoV-2 infection tests by RT-qPCR including sensitivity and specificity, and thus should be carefully considered
[83][84][85][86][83,84,85,86]. The combined swab is largely recommended as a more appropriate specimen for diagnosis by RT-qPCR
[87][88][89][87,88,89].
(6) Sensitivity: The conserved regions, ORF 1ab (RNA-dependent RNA polymerase, RdRp), envelope (E), and nucleocapsid (N) genes of SARS-CoV-2, are usually selected as the standard target genes for primer and probe design
[90][91][90,91]. However, initial reports of SARS-CoV-2 and other coronavirus sequences gave rise to an incorrect degenerate base that did not align with the SARS-CoV-2 RNA sequence found, and there were reports regarding the decreased sensitivity of using RdRp as a target gene for RT-qPCR assays
[90][92][90,92]. As the pandemic continues, many laboratories around the world rely on routine diagnostic primers and probes. Thus, proper assays can increase the sensitivity of SARS-CoV-2 detection and help prevent the further spread of the virus
[92][93][94][95][92,93,94,95].
(7) Pooling technologies: The pooling of multiple swab samples before RNA isolation and RT-qPCR analysis has been proposed as a promising solution to reduce costs and time as well as elevate the throughput of SARS-CoV-2 tests for large-scale testing as in the case of schools
[96][97][98][99][96,97,98,99]. For example, batch testing of over 100,000 hospital-collected nasopharyngeal swab samples from patients alleviated three quarters of testing reactions with a minor reduction in sensitivity, indicating the effectiveness of the pooling approach in the field
[100][101][100,101]. Current studies suggest that the pooling of individual samples before testing should be considered to increase the reliability of SARS-CoV-2 testing throughput.
Once all practical considerations have been evaluated, there are two ways that RT-qPCR can be performed. The two-step RT-qPCR method is required to convert RNA into complementary DNA (cDNA)
[102]. On the other hand, the one-step RT-qPCR method combines reverse transcription and PCR in a single tube and uses reverse transcriptase as well as a DNA polymerase
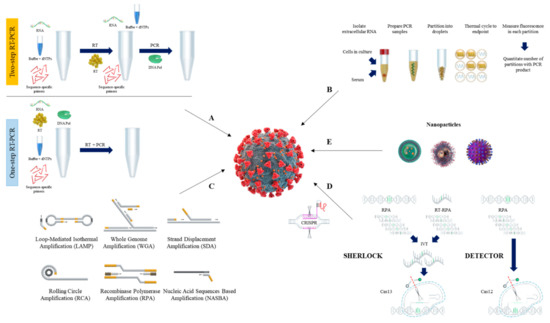
[103]. The schematic procedure of RT-qPCR is shown in A.
Figure 1. Overview of nucleic acid testing for SARS-CoV-2. The schematic procedure of RT-qPCR (A), and dPCR (B). Current isothermal amplification methods (C), CRISPR detection systems (D), and nanoparticles (E) are also shown.
A critical need for rapid and accurate diagnostic methods has emerged in the clinic and public health organizations. Several PCR-based assays have been developed and are currently being used in clinical, research, and public health laboratories
[104][105][106][104,105,106]. However, it is not clear which PCR condition they should adopt or whether the data are comparable. In response to the growing need and the lack of publicly available information, several research groups have optimized real-time PCR-based primer sets, protocols, and PCR conditions
[107][108][107,108].
Independent evaluations of the designed primer–probe sets used in SARS-CoV-2 RT–qPCR detection assays are necessary to compare and select appropriate assays
[90][109][90,109]. Additionally, several studies have utilized serum and stool specimens for the RT-qPCR-based detection method
[110][111][112][113][110,111,112,113].
2.2. Reverse Transcription Digital PCR (RT-dPCR) Method
In recent years, we have seen the advance of digital PCR (dPCR) as a complementary approach for measuring nucleic acids, a technique that is highly accurate and reproducible when targeting the viral genes of SARS-CoV-2
[114][115][116][117][114,115,116,117]. The advantages of digital PCR compared to quantitative PCR include quantification without the need for calibration curves, higher accuracy, and sensitivity that may arise from sub-optimal amplification efficacy because dPCR can detect low amounts of nucleic acid
[118][119][118,119]. The schematic procedure of dPCR is shown in B.
Reverse transcriptase quantitative PCR (RT-qPCR) and digital PCR (dPCR) have been widely used for quantitative analyses of clinical samples. Recently, many groups have developed a reverse transcription droplet digital PCR (RT-ddPCR) assay for sensitive detection of the SARS-CoV-2 virus
[120][121][122][123][120,121,122,123]. Optimization of the primer. and probe assays is necessary to remove false negatives or positives for clinical diagnosis of viral infection
[72][124][72,124]. Multiple molecular diagnostic kits have been developed and validated for use nationwide
[125]. However, the analytical sensitivity and the relative sensitivity of different kits to detect low copy number of SARS-CoV-2 viral RNA are variable
[126][127][126,127].
3. Isothermal Nucleic Acid Amplification Methods
Although the RT-qPCR method is considered the ‘gold standard’ for SARS-CoV-2 detection
[128], its limitations have stimulated the development of simple, rapid yet sensitive nucleic acid detection methods
[129]. As a result, isothermal nucleic acid amplification has emerged as an alternative detection method for SARS-CoV-2 viral RNA from clinical samples
[130]. In general, isothermal amplification techniques increase the analytical signal by increasing the target nucleic acid concentration through enzymatic activities at a fixed temperature, and simultaneously detecting the signal with colorimetric or fluorescence indicators
[131]. Changes in color, fluorescence level, or turbidity indicate the presence of SARS-CoV-2 RNA or DNA
[131]. Therefore, unlike RT-qPCR, isothermal amplification methods do not require thermal cycling instruments or specialized technicians for disease diagnosis
[132]. Current isothermal nucleic acid amplification methods used for SARS-CoV-2 detection include, but are not limited to, loop-mediated isothermal amplification (LAMP), recombinase polymerase amplification (RPA), nucleic acid sequence-based amplification (NASBA), strand-displacement amplification (SDA), and rolling circle amplification (RCA)
[133][134][135][136][137][133,134,135,136,137] (C). Herein, we describe the general procedures and components of isothermal amplification methods commonly used for diagnosis of SARS-CoV-2.
3.1. Loop-Mediated Isothermal Amplification (LAMP)
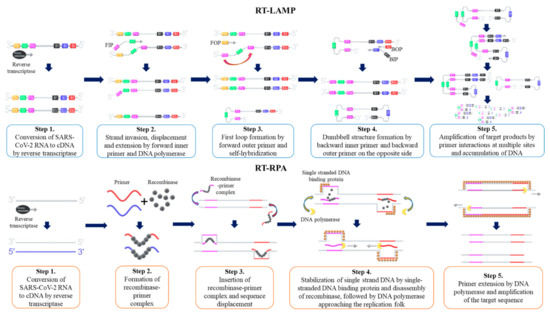
The loop-mediated isothermal amplification method, coupled with reverse transcription (RT-LAMP), is the most widely used isothermal amplification technique for SARS-CoV-2 nucleic acid detection. First described by Notomi et al.
[138], this method uses strand displacement activity of DNA polymerase and a set of inner and outer primers (four or six specific primer sequences) to amplify the target nucleic acids. LAMP is carried out at a single temperature between 60 and 65 °C, and generates up to 10
9 copies of DNA in less than an hour
[133][137][139][133,137,139]. The LAMP procedure is initiated by hybridization of the forward inner primer (FIP) toward the target DNA template, which synthesizes the complementary strand. Then, the outer primer hybridizes to the target DNA, which initiates DNA synthesis by strand displacement. Subsequently, a FIP-hybridized complementary strand is released and forms a loop structure at one end of the sequence. The corresponding sequence becomes the template for the backward inner primer (BIP), which initiates another DNA synthesis by strand displacement, and then produces a ‘dumb-bell’ like DNA structure. Self-primed DNA synthesis of the corresponding sequence then converts the ‘dumb-bell’ like structure into a ‘stem-loop’ like DNA structure. Corresponding stem-loop DNA then becomes the template for LAMP cycling, and the target DNA sequence exponentially amplifies until the reaction is completed
[138]. Amplified products are detected by changes of color as the accumulation of DNA changes, pH levels, or by changes in turbidity as magnesium pyrophosphate level increases
[140][141][142][140,141,142]. Amplified products are also detected by Calcein fluorescent dye or fluorescent intercalating dye
[129][139][129,139]. The schematic procedure of RT-LAMP is shown as .
Figure 2. Schematic procedure of reverse transcription loop-mediated isothermal amplification (RT-LAMP) and reverse transcription recombinase polymerase amplification (RT-RPA). FIP = Forward Inner Primer, FOP = Forward Outer Primer, BIP = Backward Inner Primer, BOP = Backward Outer Primer.
Researchers have made efforts to optimize RT-LAMP for the development of rapid and sensitive detection of SARS-CoV-2. Several studies have evaluated the experimental parameters for RT-LAMP such as incubation temperature, incubation time, LoD, target genes, and primer sequences
[143][144][145][143,144,145]. Aside from optimizing the experimental parameters, researchers have developed modified RT-LAMP procedures including methods without prior RNA extraction steps and high-throughput colorimetric assay methods using a 96-well plate format
[146][147][146,147]. Modified RT-LAMP procedures also include methods coupled with Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR) technology, a nanoparticle-based biosensor, and artificial intelligence
[148][149][150][151][148,149,150,151].
3.2. Recombinase Polymerase Amplification (RPA)
Recombinase polymerase amplification is another isothermal amplification method that is widely used for SARS-CoV-2 detection. First described by Piepenburg et al.
[152], RPA uses a complex of recombinase and two target specific primers (forward and reverse primers) to amplify the target nucleic acids
[152]. Once the target nucleic acids are identified, recombinase-primer complex unwinds the target DNA and allows forward and reverse primers to hybridize
[153]. The displaced DNA strand is amplified in the presence of DNA polymerase as primers elongate, and the template DNA is exponentially amplified until the reaction is completed
[153]. RPA reaction is carried out at a single temperature between 37 and 42 °C, and the reaction is completed when ATPs are depleted, typically in less than an hour
[154]. Amplified products are detected by gel electrophoresis, antigenic tags on primers and tag-specific antibodies, or fluorescent signals produced by a conjugated fluorophore and quencher on primers
[152][153][154][152,153,154]. The schematic procedure of reverse transcription RPA (RT-RPA) is shown as .
For SARS-CoV-2 detection, researchers have optimized RPA procedures by testing various experimental parameters that can now detect less than five viral copies from patient samples within 45 min from the sample collection
[134]. RPA methods have also been optimized for SARS-CoV-2 detection by coupling RPA-based amplification with various CRISPR-based detection methods
[155][156][157][155,156,157].
3.3. Other Isothermal Nuleic Acid Amplification Methods
Other than LAMP and RPA, isothermal amplification methods such as NASBA, SDA, and RCA have been used for the detection of SARS-CoV-2
[136][137][158][136,137,158]. Although we will not describe each technique in detail here, presents the general features and components of each isothermal amplification method.
Table 3. General features of the isothermal amplification techniques for SARS-CoV-2 detection.
| Method |
Components |
Temperature |
Time |
Detection Method |
Advantages * |
Disadvantages * |
| Loop-mediated isothermal amplification (LAMP) |
DNA polymerase, forward inner primer, backward inner primer, forward outer primer, backward outer primer |
60–65 °C |
>1 h |
Colorimetric, turbimetric, fluorescence probe, intercalating dye |
High specificity. Less sensitive to inhibitors in biological samples |
False positive in negative control |
| Recombinase polymerase amplification (RPA) |
Recombinase, single stranded binding protein, DNA polymerase, forward primer, reverse primer |
37–42 °C |
>1 h |
Fluorescence, antigenic-tag (antibody) |
Performed in the presence of PCR inhibitors. Fast and sensitive |
Inhibited by detergents (SDS and CTAB). Non-specific/high background signal |
| Nucleic acid sequence-based amplification (NASBA) |
RNase H, reverse transcriptase, T7 DNA-dependent RNA polymerase, forward primer with T7 promoter sequence, reverse primer |
41 °C |
>2 h |
Fluorescence |
More sensitive and less time-consuming |
Non-specific reactions/false positives |
| Strand-displacement amplification (SDA) |
DNA polymerase, restriction endonuclease, primers, dCTP, dTTP, dGTP, dATPα |
37–49 °C |
>2 h |
Fluorescence |
High specificity. Detection of large RNA molecules |
Non-specific reaction/high background signal |
| Rolling circle amplification (RCA) |
DNA ligase, DNA polymerase, primer, padlock probe |
30–37 °C |
>1.5 h |
Fluorescence |
High specificity |
False negatives and false positives |