With advancing aging, a decline in physical abilities occurs, leading to reduced mobility and loss of independence. Although many factors contribute to the physio-pathological effects of aging, an important event seems to be related to the compromised integrity of the neuromuscular system, which connects the brain and skeletal muscles via motoneurons and the neuromuscular junctions (NMJs). NMJs undergo severe functional, morphological, and molecular alterations during aging and ultimately degenerate. The effect of this decline is an inexorable decrease in skeletal muscle mass and strength, a condition generally known as sarcopenia. Moreover, several studies have highlighted how the age-related alteration of reactive oxygen species (ROS) homeostasis can contribute to changes in the neuromuscular junction morphology and stability, leading to the reduction in fiber number and innervation. Increasing evidence supports the involvement of epigenetic modifications in age-dependent alterations of the NMJ. In particular, DNA methylation, histone modifications, and miRNA-dependent gene expression represent the major epigenetic mechanisms that play a crucial role in NMJ remodeling. It is established that environmental and lifestyle factors, such as physical exercise and nutrition that are susceptible to change during aging, can modulate epigenetic phenomena and attenuate the age-related NMJs changes.
- aging
- ROS
- oxidative stress
- ALS
- skeletal muscle
- nutrition
- exercise
Note: The following contents are extract from your paper. The entry will be online only after author check and submit it.
1. Introduction
2. Age-Related Changes in NMJ
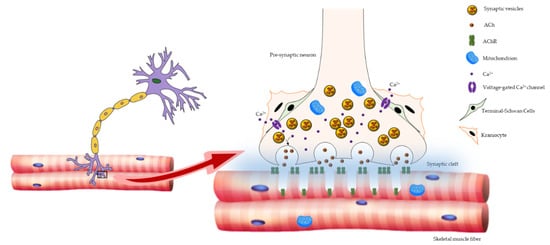
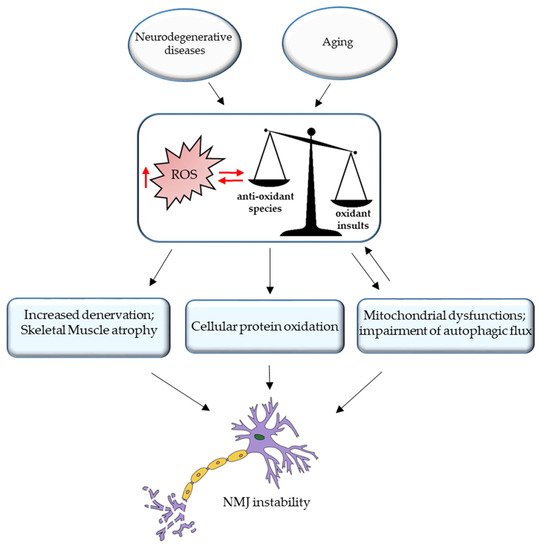
3. ROS and NMJ Degeneration
References
- Chhetri, J.K.; de Souto Barreto, P.; Fougère, B.; Rolland, Y.; Vellas, B.; Cesari, M. Chronic inflammation and sarcopenia: A regenerative cell therapy perspective. Exp. Gerontol. 2018, 103, 115–123.
- Dickinson, J.M.; Volpi, E.; Rasmussen, B.B. Exercise and Nutrition to Target Protein Synthesis Impairments in Aging Skeletal Muscle. Exerc. Sport Sci. Rev. 2013, 41, 216–223.
- Marzetti, E.; Calvani, R.; Cesari, M.; Buford, T.W.; Lorenzi, M.; Behnke, B.J.; Leeuwenburgh, C. Mitochondrial dysfunction and sarcopenia of aging: From signaling pathways to clinical trials. Int. J. Biochem. Cell Biol. 2013, 45, 2288–2301.
- Sousa-Victor, P.; Muñoz-Cánoves, P. Regenerative decline of stem cells in sarcopenia. Mol. Aspects Med. 2016, 50, 109–117.
- Larsson, L.; Degens, H.; Li, M.; Salviati, L.; Lee, Y.I.; Thompson, W.; Kirkland, J.L.; Sandri, M. Sarcopenia: Aging-related loss of muscle mass and function. Physiol. Rev. 2019, 99, 427–511.
- Hepple, R.T.; Rice, C.L. Innervation and neuromuscular control in ageing skeletal muscle. J. Physiol. 2016, 594, 1965–1978.
- Wu, R.; De Vito, G.; Delahunt, E.; Ditroilo, M. Age-related Changes in Motor Function (I)Mechanical and Neuromuscular Factors. Int. J. Sports Med. 2020, 41, 709–719.
- Rudolf, R.; Khan, M.M.; Witzemann, V. Motor Endplate—Anatomical, Functional, and Molecular Concepts in the Historical Perspective. Cells 2019, 8, 387.
- Mège, R.M.; Goudou, D.; Giaume, C.; Nicolet, M.; Rieger, F. Is intercellular communication via Gap junctions required for myoblast fusion?? Cell Commun. Adhes. 1994, 2, 329–343.
- Schiaffino, S.; Reggiani, C. Molecular diversity of myofibrillar proteins: Gene regulation and functional significance. Physiol. Rev. 1996, 76, 371–423.
- Hughes, S.M. Muscle development: Electrical control of gene expression. Curr. Biol. 1998, 8, R892–R894.
- Olson, E.N.; Williams, R.S. Calcineurin signaling and muscle remodeling. Cell 2000, 101, 689–692.
- Buller, A.J.; Eccles, J.C.; Eccles, R.M. Differentiation of fast and slow muscles in the cat hind limb. J. Physiol. 1960, 150, 399–416.
- Pette, D.; Vrbová, G. Invited review: Neural control of phenotypic expression in mammalian muscle fibers. Muscle Nerve 1985, 8, 676–689.
- Klitgaard, H.; Zhou, M.; Schiaffino, S.; Betto, R.; Salviati, G.; Saltin, B. Ageing alters the myosin heavy chain composition of single fibres from human skeletal muscle. Acta Physiol. Scand. 1990, 140, 55–62.
- Choi, S.-J. Age-related functional changes and susceptibility to eccentric contraction-induced damage in skeletal muscle cell. Integr. Med. Res. 2016, 5, 171–175.
- Bao, Z.; Cui, C.; Chow, S.K.H.; Qin, L.; Wong, R.M.Y.; Cheung, W.H. AChRs Degeneration at NMJ in Aging-Associated Sarcopenia–A Systematic Review. Front. Aging Neurosci. 2020, 12, 597811.
- Vaughan, D.W. Effects of advancing age on peripheral nerve regeneration. J. Comp. Neurol. 1992, 323, 219–237.
- Delmonico, M.J.; Harris, T.B.; Visser, M.; Park, S.W.; Conroy, M.B.; Velasquez-Mieyer, P.; Boudreau, R.; Manini, T.M.; Nevitt, M.; Newman, A.B.; et al. Longitudinal study of muscle strength, quality, and adipose tissue infiltration. Am. J. Clin. Nutr. 2009, 90, 1579–1585.
- Lexell, J.; Taylor, C.C.; Sjöström, M. What is the cause of the ageing atrophy?. Total number, size and proportion of different fiber types studied in whole vastus lateralis muscle from 15- to 83-year-old men. J. Neurol. Sci. 1988, 84, 275–294.
- Wokke, J.H.J.; Jennekens, F.G.I.; van den Oord, C.J.M.; Veldman, H.; Smit, L.M.E.; Leppink, G.J. Morphological changes in the human end plate with age. J. Neurol. Sci. 1990, 95, 291–310.
- Rowan, S.L.; Rygiel, K.; Purves-Smith, F.M.; Solbak, N.M.; Turnbull, D.M.; Hepple, R.T. Denervation causes fiber atrophy and myosin heavy chain co-expression in senescent skeletal muscle. PLoS ONE 2012, 7, 29082.
- Valdez, G.; Tapia, J.C.; Kang, H.; Clemenson, G.D.; Gage, F.H.; Lichtman, J.W.; Sanes, J.R. Attenuation of age-related changes in mouse neuromuscular synapses by caloric restriction and exercise. Proc. Natl. Acad. Sci. USA 2010, 107, 14863–14868.
- Cerrato, F.; Sparago, A.; Ariani, F.; Brugnoletti, F.; Calzari, L.; Coppedè, F.; De Luca, A.; Gervasini, C.; Giardina, E.; Gurrieri, F.; et al. DNA methylation in the diagnosis of monogenic diseases. Genes 2020, 11, 355.
- Bennett, S.A.; Tanaz, R.; Cobos, S.N.; Torrente, M.P. Epigenetics in amyotrophic lateral sclerosis: A role for histone post-translational modifications in neurodegenerative disease. Transl. Res. 2019, 204, 19–30.
- Pratt, J.; De Vito, G.; Narici, M.; Boreham, C. Neuromuscular Junction Aging: A Role for Biomarkers and Exercise. J. Gerontol. A Biol. Sci. Med. Sci. 2021, 76, 576–585.
- Lepore, E.; Casola, I.; Dobrowolny, G.; Musarò, A. Neuromuscular Junction as an Entity of Nerve-Muscle Communication. Cells 2019, 8, 906.
- Ruegg, M.A. Organization of synaptic myonuclei by Syne proteins and their role during the formation of the nerve-muscle synapse. Proc. Natl. Acad. Sci. USA 2005, 102, 5643–5644.
- Li, L.; Xiong, W.C.; Mei, L. Neuromuscular Junction Formation, Aging, and Disorders. Annu. Rev. Physiol. 2018, 80, 159–188.
- Schaeffer, L.; De Kerchove D’Exaerde, A.; Changeux, J.P. Targeting transcription to the neuromuscular synapse. Neuron 2001, 31, 15–22.
- Saha, R.N.; Pahan, K. HATs and HDACs in neurodegeneration: A tale of disconcerted acetylation homeostasis. Cell Death Differ. 2006, 13, 539–550.
- Battey, E.; Stroud, M.J.; Ochala, J. Using nuclear envelope mutations to explore age-related skeletal muscle weakness. Clin. Sci. 2020, 134, 2177–2187.
- Zhang, X.; Xu, R.; Zhu, B.; Yang, X.; Ding, X.; Duan, S.; Xu, T.; Zhuang, Y.; Han, M. Syne-1 and Syne-2 play crucial roles in myonuclear anchorage and motor neuron innervation. Development 2007, 134, 901–908.
- Grady, R.M.; Starr, D.A.; Ackerman, G.L.; Sanes, J.R.; Han, M. Syne proteins anchor muscle nuclei at the neuromuscular junction. Proc. Natl. Acad. Sci. USA 2005, 102, 4359–4364.
- Burke, B.; Stewart, C.L. The laminopathies: The functional architecture of the nucleus and its contribution to disease. Annu. Rev. Genomics Hum. Genet. 2006, 7, 369–405.
- Burke, B.; Stewart, C.L. The nuclear lamins: Flexibility in function. Nat. Rev. Mol. Cell Biol. 2013, 14, 13–24.
- Hutchison, C.J. Lamins: Building blocks or regulators of gene expression? Nat. Rev. Mol. Cell Biol. 2002, 3, 848–858.
- Mounkes, L.; Kozlov, S.; Burke, B.; Stewart, C.L. The laminopathies: Nuclear structure meets disease. Curr. Opin. Genet. Dev. 2003, 13, 223–230.
- Broers, J.L.V.; Ramaekers, F.C.S.; Bonne, G.; Ben Yaou, R.; Hutchison, C.J. Nuclear lamins: Laminopathies and their role in premature ageing. Physiol. Rev. 2006, 86, 967–1008.
- De Sandre-Giovannoli, A.; Chaouch, M.; Kozlov, S.; Vallat, J.M.; Tazir, M.; Kassouri, N.; Szepetowski, P.; Hammadouche, T.; Vandenberghe, A.; Stewart, C.L.; et al. Homozygous defects in LMNA, encoding lamin A/C nuclear-envelope proteins, cause autosomal recessive axonal neuropathy in human (Charcot-Marie-Tooth disorder type 2) and mouse. Am. J. Hum. Genet. 2002, 70, 726–736.
- Scaffidi, P.; Misteli, T. Lamin A-dependent nuclear defects in human aging. Science 2006, 312, 1059–1063.
- Gao, N.; Zhao, K.; Cao, Y.; Ren, X.; Jing, H.; Xing, G.; Xiong, W.C.; Mei, L. A role of lamin A/C in preventing neuromuscular junction decline in mice. J. Neurosci. 2020, 40, 7203–7215.
- Coppedè, F. Epigenetics of neuromuscular disorders. Epigenomics 2020, 12, 2125–2139.
- Azpurua, J.; Eaton, B.A. Neuronal epigenetics and the aging synapse. Front. Cell. Neurosci. 2015, 9.
- Latcheva, N.K.; Viveiros, J.M.; Waddell, E.A.; Nguyen, P.T.T.; Liebl, F.L.W.; Marenda, D.R. Epigenetic crosstalk: Pharmacological inhibition of HDACs can rescue defective synaptic morphology and neurotransmission phenotypes associated with loss of the chromatin reader Kismet. Mol. Cell. Neurosci. 2018, 87, 77–85.
- Osseni, A.; Ravel-Chapuis, A.; Thomas, J.-L.; Gache, V.; Schaeffer, L.; Jasmin, B.J. HDAC6 regulates microtubule stability and clustering of AChRs at neuromuscular junctions. J. Cell Biol. 2020, 219.
- Lu, L.; Liu, Y.; Liu, Y.; Zhang, F.; Wang, H.; Zhang, Q.; Pan, F. Secreted miRNAs in the tripartite neuromuscular junction. ExRNA 2019, 1, 1–4.
- Castro, R.; Taetzsch, T.; Vaughan, S.K.; Godbe, K.; Chappell, J.; Settlage, R.E.; Valdez, G. Specific labeling of synaptic schwann cells reveals unique cellular and molecular features. Elife 2020, 9, 1–19.
- Feng, Z.; Ko, C.P. Schwann cells promote synaptogenesis at the neuromuscular junction via transforming growth factor-β1. J. Neurosci. 2008, 28, 9599–9609.
- Sugiura, Y.; Lin, W. Neuron-glia interactions: The roles of Schwann cells in neuromuscular synapse formation and function. Biosci. Rep. 2011, 31, 295–302.
- Court, F.A.; Gillingwater, T.H.; Melrose, S.; Sherman, D.L.; Greenshields, K.N.; Morton, A.J.; Harris, J.B.; Willison, H.J.; Ribchester, R.R. Identity, developmental restriction and reactivity of extralaminar cells capping mammalian neuromuscular junctions. J. Cell Sci. 2008, 121, 3901–3911.
- Gonzalez-Freire, M.; de Cabo, R.; Studenski, S.A.; Ferrucci, L. The neuromuscular junction: Aging at the crossroad between nerves and muscle. Front. Aging Neurosci. 2014, 6, 208.
- Rudolf, R.; Khan, M.M.; Labeit, S.; Deschenes, M.R. Degeneration of neuromuscular junction in age and dystrophy. Front. Aging Neurosci. 2014, 6, 99.
- Willadt, S.; Nash, M.; Slater, C. Age-related changes in the structure and function of mammalian neuromuscular junctions. Ann. N. Y. Acad. Sci. 2018, 1412, 41–53.
- Deschenes, M.R.; Hurst, T.E.; Ramser, A.E.; Sherman, E.G. Presynaptic to postsynaptic relationships of the neuromuscular junction are held constant across age and muscle fiber type. Dev. Neurobiol. 2013, 73, 744–753.
- Bowen, D.C.; Park, J.S.; Bodine, S.; Stark, J.L.; Valenzuela, D.M.; Stitt, T.N.; Yancopoulos, G.D.; Lindsay, R.M.; Glass, D.J.; Distefano, P.S. Localization and regulation of MuSK at the neuromuscular junction. Dev. Biol. 1998, 199, 309–319.
- Itou, Y.; Nochi, R.; Kuribayashi, H.; Saito, Y.; Hisatsune, T. Cholinergic activation of hippocampal neural stem cells in aged dentate gyrus. Hippocampus 2011, 21, 446–459.
- Taetzsch, T.; Valdez, G. NMJ maintenance and repair in aging. Curr. Opin. Physiol. 2018, 4, 57–64.
- Jang, Y.C.; Van Remmen, H. Age-associated alterations of the neuromuscular junction. Exp. Gerontol. 2011, 46, 193–198.
- Kang, H.; Tian, L.; Mikesh, M.; Lichtman, J.W.; Thompson, W.J. Terminal schwann cells participate in neuromuscular synapse remodeling during reinnervation following nerve injury. J. Neurosci. 2014, 34, 6323–6333.
- Barik, A.; Lu, Y.; Sathyamurthy, A.; Bowman, A.; Shen, C.; Li, L.; Xiong, W.C.; Mei, L. LRP4 is critical for neuromuscular junction maintenance. J. Neurosci. 2014, 34, 13892–13905.
- Ohno, K.; Ohkawara, B.; Ito, M. Agrin-LRP4-MuSK signaling as a therapeutic target for myasthenia gravis and other neuromuscular disorders. Expert Opin. Ther. Targets 2017, 21, 949–958.
- DeChiara, T.M.; Bowen, D.C.; Valenzuela, D.M.; Simmons, M.V.; Poueymirou, W.T.; Thomas, S.; Kinetz, E.; Compton, D.L.; Rojas, E.; Park, J.S.; et al. The receptor tyrosine kinase MuSK is required for neuromuscular junction formation in vivo. Cell 1996, 85, 501–512.
- Bowen, D.C.; Sugiyama, J.; Ferns, M.; Hall, Z.W. Neural agrin activates a high-affinity receptor in C2 muscle cells that is unresponsive to muscle agrin. J. Neurosci. 1996, 16, 3791–3797.
- Ferns, M.J.; Campanelli, J.T.; Hoch, W.; Scheller, R.H.; Hall, Z. The ability of agrin to cluster AChRs depends on alternative splicing and on cell surface proteoglycans. Neuron 1993, 11, 491–502.
- Gesemann, M.; Denzer, A.J.; Ruegg, M.A. Acetylcholine receptor-aggregating activity of agrin isoforms and mapping of the active site. J. Cell Biol. 1995, 128, 625–636.
- Lin, W.; Burgess, R.W.; Dominguez, B.; Pfaff, S.L.; Sanes, J.R.; Lee, K.F. Distinct roles of nerve and muscle in postsynaptic differentiation of the neuromuscular synapse. Nature 2001, 410, 1057–1064.
- Yang, J.F.; Cao, G.; Koirala, S.; Reddy, L.V.; Ko, C.P. Schwann cells express active agrin and enhance aggregation of acetylcholine receptors on muscle fibers. J. Neurosci. 2001, 21, 9572–9584.
- Bütikofer, L.; Zurlinden, A.; Bolliger, M.F.; Kunz, B.; Sonderegger, P. Destabilization of the neuromuscular junction by proteolytic cleavage of agrin results in precocious sarcopenia. FASEB J. 2011, 25, 4378–4393.
- Ibebunjo, C.; Chick, J.M.; Kendall, T.; Eash, J.K.; Li, C.; Zhang, Y.; Vickers, C.; Wu, Z.; Clarke, B.A.; Shi, J.; et al. Genomic and Proteomic Profiling Reveals Reduced Mitochondrial Function and Disruption of the Neuromuscular Junction Driving Rat Sarcopenia. Mol. Cell. Biol. 2013, 33, 194–212.
- Dalkin, W.; Taetzsch, T.; Valdez, G. The fibular nerve injury method: A reliable assay to identify and test factors that repair neuromuscular junctions. J. Vis. Exp. 2016, 2016, e54186.
- Zhou, J.; Yi, J.; Fu, R.; Liu, E.; Siddique, T.; Rios, E.; Deng, H.X. Hyperactive intracellular calcium signaling associated with localized mitochondrial defects in skeletal muscle of an animal model of amyotrophic lateral sclerosis. J. Biol. Chem. 2010, 285, 705–712.
- Muller, F.L.; Song, W.; Jang, Y.C.; Liu, Y.; Sabia, M.; Richardson, A.; Van Remmen, H. Denervation-induced skeletal muscle atrophy is associated with increased mitochondrial ROS production. Am. J. Physiol. Integr. Comp. Physiol. 2007, 293, 1159–1168.
- Banker, B.Q.; Kelly, S.S.; Robbins, N. Neuromuscular transmission and correlative morphology in young and old mice. J. Physiol. 1983, 339, 355–377.
- Dobrowolny, G.; Martini, M.; Scicchitano, B.M.; Romanello, V.; Boncompagni, S.; Nicoletti, C.; Pietrangelo, L.; De Panfilis, S.; Catizone, A.; Bouchè, M.; et al. Muscle Expression of SOD1 G93A Triggers the Dismantlement of Neuromuscular Junction via PKC-Theta. Antioxid. Redox Signal. 2018, 28, 1105–1119.
- Rocha, M.C.; Pousinha, P.A.; Correia, A.M.; Sebastião, A.M.; Ribeiro, J.A. Early Changes of Neuromuscular Transmission in the SOD1(G93A) Mice Model of ALS Start Long before Motor Symptoms Onset. PLoS ONE 2013, 8.
- Scialò, F.; Fernández-Ayala, D.J.; Sanz, A. Role of Mitochondrial Reverse Electron Transport in ROS Signaling: Potential Roles in Health and Disease. Front. Physiol. 2017, 8, 428.
- Li, A.; Yi, J.; Li, X.; Zhou, J. Physiological Ca2+ Transients Versus Pathological Steady-State Ca2+ Elevation, Who Flips the ROS Coin in Skeletal Muscle Mitochondria. Front. Physiol. 2020, 11.
- Hyatt, H.W.; Powers, S.K. Mitochondrial dysfunction is a common denominator linking skeletal muscle wasting due to disease, aging, and prolonged inactivity. Antioxidants 2021, 10, 588.
- Simon, H.U.; Haj-Yehia, A.; Levi-Schaffer, F. Role of reactive oxygen species (ROS) in apoptosis induction. Apoptosis 2000, 5, 415–418.
- Zhang, J.; Wang, X.; Vikash, V.; Ye, Q.; Wu, D.; Liu, Y.; Dong, W. ROS and ROS-Mediated Cellular Signaling. Oxid. Med. Cell. Longev. 2016, 2016, 1–18.
- Scicchitano, B.M.; Pelosi, L.; Sica, G.; Musarò, A. The physiopathologic role of oxidative stress in skeletal muscle. Mech. Ageing Dev. 2018, 170, 37–44.
- Dupuis, L.; Gonzalez de Aguilar, J.L.; Echaniz-Laguna, A.; Eschbach, J.; Rene, F.; Oudart, H.; Halter, B.; Huze, C.; Schaeffer, L.; Bouillaud, F.; et al. Muscle mitochondrial uncoupling dismantles neuromuscular junction and triggers distal degeneration of motor neurons. PLoS ONE 2009, 4, e5390.
- Fischer, L.R.; Li, Y.; Asress, S.A.; Jones, D.P.; Glass, J.D. Absence of SOD1 leads to oxidative stress in peripheral nerve and causes a progressive distal motor axonopathy. Exp. Neurol. 2012, 233, 163–171.
- Fischyer, L.R.; Culver, D.G.; Tennant, P.; Davis, A.A.; Wang, M.; Castellano-Sanchez, A.; Khan, J.; Polak, M.A.; Glass, J.D. Amyotrophic lateral sclerosis is a distal axonopathy: Evidence in mice and man. Exp. Neurol. 2004, 185, 232–240.
- Gurney, M.E.; Pu, H.; Chiu, A.Y.; Dal Canto, M.C.; Polchow, C.Y.; Alexander, D.D.; Caliendo, J.; Hentati, A.; Kwon, Y.W.; Deng, H.X.; et al. Motor neuron degeneration in mice that express a human Cu, Zn superoxide dismutase mutation. Science 1994, 264, 1772–1775.
- Hayes, L.R.; Asress, S.A.; Li, Y.; Galkin, A.; Stepanova, A.; Kawamata, H.; Manfredi, G.; Glass, J.D. Distal denervation in the SOD1 knockout mouse correlates with loss of mitochondria at the motor nerve terminal. Exp. Neurol. 2019, 318, 251–257.
- Pollari, E.; Goldsteins, G.; Bart, G.; Koistinaho, J.; Giniatullin, R. The role of oxidative stress in degeneration of the neuromuscular junction in amyotrophic lateral sclerosis. Front. Cell. Neurosci. 2014, 8, 131.
- Martin, L.J.; Wong, M. Skeletal Muscle-Restricted Expression of Human SOD1 in Transgenic Mice Causes a Fatal ALS-Like Syndrome. Front. Neurol. 2020, 11.
- Wong, M.; Martin, L.J. Skeletal muscle-restricted expression of human SOD1 causes motor neuron degeneration in transgenic mice. Hum. Mol. Genet. 2010, 19, 2284–2302.
- Ji, L.L.; Yeo, D.; Kang, C.; Zhang, T. The role of mitochondria in redox signaling of muscle homeostasis. J. Sport Heal. Sci. 2020, 9, 386–393.
- Giniatullin, A.; Petrov, A.; Giniatullin, R. The involvement of P2Y12 receptors, NADPH oxidase, and lipid rafts in the action of extracellular ATP on synaptic transmission at the frog neuromuscular junction. Neuroscience 2015, 285, 324–332.
- Jackson, M.J.; Vasilaki, A.; McArdle, A. Cellular mechanisms underlying oxidative stress in human exercise. Free Radic. Biol. Med. 2016, 98, 13–17.
- Gomez-Cabrera, M.C.; Domenech, E.; Viña, J. Moderate exercise is an antioxidant: Upregulation of antioxidant genes by training. Free Radic. Biol. Med. 2008, 44, 126–131.
- Wright, V.P.; Reiser, P.J.; Clanton, T.L. Redox modulation of global phosphatase activity and protein phosphorylation in intact skeletal muscle. J. Physiol. 2009, 587, 5767–5781.
- Bejma, J.; Ji, L.L. Rapid communication aging and acute exercise enhance free radical generation in rat skeletal muscle. J. Appl. Physiol. 1999, 87, 465–470.
- Damiano, S.; Muscariello, E.; La Rosa, G.; Di Maro, M.; Mondola, P.; Santillo, M. Dual role of reactive oxygen species in muscle function: Can antioxidant dietary supplements counteract age-related sarcopenia? Int. J. Mol. Sci. 2019, 20, 3815.
- Sinha, S.; Ray, U.S.; Saha, M.; Singh, S.N.; Tomar, O.S. Antioxidant and redox status after maximal aerobic exercise at high altitude in acclimatized lowlanders and native highlanders. Eur. J. Appl. Physiol. 2009, 106, 807–814.
- Sinha, S.; Singh, S.N.; Saha, M.; Kain, T.C.; Tyagi, A.K.; Ray, U.S. Antioxidant and oxidative stress responses of sojourners at high altitude in different climatic temperatures. Int. J. Biometeorol. 2010, 54, 85–92.
- Carnio, S.; LoVerso, F.; Baraibar, M.A.; Longa, E.; Khan, M.M.; Maffei, M.; Reischl, M.; Canepari, M.; Loefler, S.; Kern, H.; et al. Autophagy Impairment in Muscle Induces Neuromuscular Junction Degeneration and Precocious Aging. Cell Rep. 2014, 8, 1509–1521.
- You, J.S.; Singh, N.; Reyes-Ordonez, A.; Khanna, N.; Bao, Z.; Zhao, H.; Chen, J. ARHGEF3 Regulates Skeletal Muscle Regeneration and Strength through Autophagy. Cell Rep. 2021, 34.