Even when the molecular structure of CD34 is well recognized, its function is far from being completely understood. In hematopoiesis, CD34 has roles of cytoadhesion and the regulation of cell differentiation. CD34
+ cells represent a proportion of the total mesenchymal stem cells (MSCs), and are associated with high colony forming efficiency and long-term proliferative capacity
[21][22][39,40]. Moreover, CD34
+ MSCs have exhibited a propensity for endothelial transdifferentiation. Thus, CD34
+/CD90
+ cells of human adipose tissue were able to form a sphere cluster and be differentiated in endothelial cells that form capillary-like structures producing a high level of VEGF
[23][41]. A diffuse cytoplasmic with perinuclear enhancement of bcl-2 staining has been constantly described in SFT
[24][42]. The level of bcl-2 positivity ranges from 70 to 86% in the largest series of SFT
[19][25][37,43]. Of note, bcl-2 immunostaining was positive in benign conditions with spindle cellularity (). Further, apart from SFT, it is expressed in some fibroblastic spindle cell sarcomas and in the spindle component of DD-LPS or in synovial sarcoma. A close relationship of CD34 staining is seen with bcl-2, being coincident in several tumors such as SFT, dermatofibrosarcoma protuberans, Kaposi sarcoma or gastro-intestinal stromal tumor (GIST), as well as in other previously mentioned benign conditions
[26][44]. In SFT, bcl-2 expression was seen regardless of the mitotic activity or the cellularity. Analyses in mammalian tissues determined that bcl-2 protein expression is common in stem cells, endocrine tissue, and long-lived cells
[27][45]. Considered together, this raises the possibility that the pathogenesis of SFT could be explained as a result of neoplastic transformation of a fibroblastic precursor CD34
+/bcl-2
+. In addition, bcl-2 expression could be induced by STAT6 through IL-4. This mechanism is physiologically activated in lymphocytes where this signaling pathway would maintain the T cells activated, avoiding apoptosis
[28][46]. The overexpression of bcl-2 in SFT could explain the chemo-resistance seen in this entity. Additionally, the bcl-2 expression detected in synovial sarcoma could be explained by the characteristic translocation t (X, 18) that would affect the bcl-2 gene allocated in chromosome 18
[29][47]. The positive expression of bcl-2 seen in neural neoplasms could be due to the fact that these tumors stem from the neural crest cell lineage, which also expresses bcl-2. The protein expression of CD99 is extensively present in SFT, showing strong membranous predominant staining, or membranous cytoplasmic, in more than 80% of cases. The glycosylated transmembrane protein CD99 is implicated in several cellular functions such as cell adhesion, migration, differentiation, endo and exocytosis among others
[30][48]. In malignancy, CD99 has been demonstrated to have a remarkable role in migration, invasion and metastasis. In this sense, CD99 has behaved as an oncogene in several tumors including some sarcomas such as Ewing sarcoma, synovial sarcoma or rhabdomyosarcoma. However, there is an increasing number of tumors in which CD99 expression is diffuse in an early stage or in benign conditions, but is lacking or reduced in an advanced stage or in the malignant counterpart
[31][32][49,50]. In this latter subset of tumors, which includes osteosarcoma, CD99 acts as a suppressor gene. In addition, two isoforms of CD99 (wild type and truncated forms) have been described with opposite functions. While CD99
wt inhibits migration, metastasis, anoikis resistance and anchorage-independent growth, the truncated form exhibits the opposite functions
[33][51]. CD99 is highly expressed in CD34
+ bone marrow cells and in leukocytes, whatever the lineage, and it is a determinant in the orientation of immune response
[34][52]. As CD99 expression is usually lost in the dedifferentiated SFT (in the DD zones), it is probable that CD99 would act also as a tumor suppressor in the context of SFT
[35][53].
In all, one might speculate that precursor cells of SFT would harbor an early expression of CD34+ and bcl-2, probably a progenitor of fibroblasts or myofibroblasts, along with CD99+ upon which other genetic early hits had been added, such as the characteristic NAB2–STAT6 transcript. The latter would ultimately facilitate proliferation through EGR1 signaling.
4. Dedifferentiated SFT (DD-SFT)
Dedifferentiation can occur at the end of the transforming histological stage of SFT, reflecting that new genetic hits have emerged in the tumor or that dedifferentiated clones have evolved from the initial malignant process, until they govern the tumor biology. This dedifferentiation process is not exclusive to SFT, rather it is observed in a wide spectrum of malignancies, such as melanomas, carcinomas or even other sarcomas (DD-LPS or chondrosarcomas, for instance). This subtype can be underestimated when core biopsies do not reach the dedifferentiated component or after a resection of bulky SFT if the sampling is not complete. DD-SFT can be diagnosed de novo or following a recurrence of indolent or aggressive SFT. Characteristically, DD-SFT is diagnosed if an abrupt area of high grade sarcomatous or anaplastic cells appears in the SFT bed. As previously mentioned, some protein expressions are frequently lost in DD-SFT, such as CD34,
[20][36][37][38,54,55] CD99
[38][56] and STAT6
[14][39][32,57]. It is not yet clear which are the mechanisms underlying the loss of expression of the previous proteins, but a kind of post-translational control through ubiquitination has been postulated, at least for the loss of STAT6 nuclear expression
[14][32]. The mutation of
TP53, and accordingly, a nuclear positive immunostaining, has been found in some high-grade SFTs and in many DD-SFTs. This finding was reported a long time ago
[20][40][38,58] and corroborated in recent times by comparative genomic hybridization, demonstrating the loss of 17p, always involving
TP53, in high-grade and DD-SFT
[14][41][42][32,59,60]. Another onco-suppressor,
RB1, is also frequently lost in DD-SFT, something supported by the disappearance of previously verified nuclear immunostaining and by the most frequent copy number abnormalities, the loss of 13q, always affecting
RB1 [14][32]. Interestingly, in the transit towards higher dedifferentiation, SFT cells display a complex cytogenetic profile with numerous copy number alterations, indicating an increase in genomic instability
[14][32]. Oxidative stress (ROS) could also contribute to this instability via EGR1, which is a transcriptional activator of
NOX4. In some disease contexts, such as diabetic kidney disease,
EGR1, NOX4 and
ROS have been found as critical components
[43][61]. Interestingly,
NOX4 is overexpressed in SFT. These genomic gains and losses are nonrandom events, likewise other dedifferentiation processes observed in sarcoma, such as DD-LPS or DD chondrosarcoma, where recurrent genomic events are detected. The critical genomic events that induce different SFT subtypes from one precursor or maybe convert low-grade SFT into high-grade SFT and then to DD-SFT, changing from
NAB2–STAT6 addiction to other genomic drivers, are just being explored from recent times.
Insulin-like growth factor 2 (IGF2) and insulin-like growth factor 2 receptor (IGF2R) are overexpressed in a substantial proportion of SFT cases, and this overexpression is detected by immunohistochemistry
[44][62]. In fact, the serum increase in IGF2 would be responsible for the hypoglycemic syndrome (Doege–Potter syndrome). Hypoglycemia has been proposed as an independent prognostic marker in SFT for a higher probability of metastatic recurrence and death
[45][63]. Along the same lines, early reports already related the appearance of hypoglycemia to a larger tumor diameter and more aggressive behavior
[46][64]. However, some controversies still remain regarding the prognostic implications and functionality of IGF2R. Additionally, the overexpression of interferon-stimulated gene 15 (
ISG15) significantly correlated with worse PFS and OS in translational research of a phase II clinical trial exploring pazopanib as first line of antiangiogenesis in advanced/metastatic SFT
[47][65].
ISG15 has been implicated in stemness, cancer survival and drug resistance
[48][66].
The presence of
TERT promoter mutation had a worse prognostic role in some series
[49][50][67,68] but not in others
[51][69]. Therefore, further investigation is needed to know its real prognostic impact.
5. Molecular Biology
The identification of
NAB2–STAT6 fusion within chromosome 12, communicated in 2013 by three different research groups, was an important milestone in terms of understanding this entity
[52][53][54][70,71,72]. According to some authors, the key dysfunction in the transcript
NAB2–STAT6 lies in the disturbed function of
NAB2, rather than
STAT6 deregulation. The reason is based on the fact that all the genomic fusions between
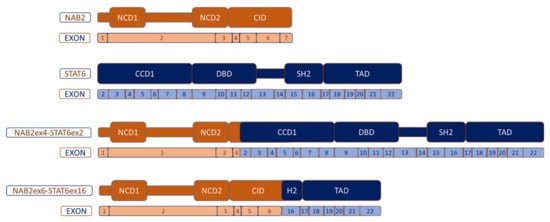
NAB2–STAT6 entail a protein transcript that exchanged at least one repressor domain from NAB2 for an activation domain from STAT6. This results in a dysregulation of early growth response (EGR) signaling. NAB2 is constituted, on the one hand, by an N-terminal binding domain (NAB2 conserved domain 1-NCD1) that interacts with EGR1, and NCD2 which is important for transcriptional repression and, on the other hand, the carboxy-terminal, which includes a chromodomain helicase DNA-binding protein 4 (CHD4) interacting domain (CID), important for transcriptional repression as well. CHD4 is a subunit of the nucleosome remodeling and histone deacetylase complex (NuRD) with enzymatic function ().
Figure 2. Most common NAB2–STAT6 fusion variants. NCD: NAB-conserved domain; CID: CHD4-interacting domain; CCD1: Coiled-coil domain 1; DBD: DNA-binding domain; SH2: Src homology 2 and TAD: transcriptional activator domain.
As the EGR1-interacting NCD1 is always present in the fusion protein, an enhancement of the expression of EGR1-target genes is expected in SFT. EGR1 is one of four cysteine-rich zinc finger transcription factors.
EGR1 expression is activated by different factors such as cytokines, hormones or growth factors such as TGF-β. This gene has been implicated in cancer, inflammation or fibrosis. Of note,
NAB2 is a target of EGR1 and
EGR1 is a target of NAB2. This latter regulation is produced by the binding of ERG1 to the R1 inhibitory domain and through CHD3 and CHD4 proteins that have enzymatic function and belong to NuRD, a remodeling chromatin complex with relevance in transcription regulation
[55][73]. As the wild type
NAB2 gene is a repressor of EGR1 transcription activity, and in the context of the
NAB2–STAT6 transcript, where the carboxi-terminal parts of
NAB2 (relevant for transcriptional repression) have been exchanged, an increase in transcription for
EGR1 target genes could be expected. However, this was not found in some investigations
[54][72]. This could be explained by the fact that
NAB2 sometimes potentiates rather than represses
EGR1 transcription. In any case, EGR1′s effects are not fully characterized. Among those EGR1 target genes found to be dysregulated in SFT, several are involved in fetal development. Thus,
HOX genes (class I homeobox genes) have been shown to be overexpressed in SFT, except for
HOXD [53][54][71,72].
Of note, NuRD also has a relevant prominence for the normal differentiation of embryonic stem cells by downregulating the expression of
ZFP42,
TBX3,
KLF4, and
KLF5 genes. Intriguingly, some of these genes are downregulated in SFT
[54][72]. In other words, in the context of SFT, NuRD’s function is to restrict these pluripotency genes. It should be questioned if in DD-SFT some of the NuRD complex functions are lost, such as the
Mbd3, since the mutation of this gene induced the overexpression of the pluripotency genes mentioned above
[56][74].
On the other hand, the
STAT6 gene is a member of the
STAT family encoding cytoplasmic transcription factors, which regulate gene expression, transmitting signals to the nucleus and binding to certain DNA promoters. The
STAT6 gene consists of 23 exons and functionally has the following structure: N-terminal, coiled-coil domain (CCD), DNA binding domain (DBD1), a linker domain (LD), a Src-homology 2 domain (SH2), a tyrosine phosphorylation site (pY) and a C-terminal transcriptional activation domain (TAD). The SH2 domain is critical for binding to a receptor (IL-4R or IL-13R) and consecutive activation of STAT6 through the phosphorylation of tyrosine residues with the intervention of Janus kinases (JAK), specifically Jak1 and Jak3. After the phosphorylation of receptors, STAT6 binds to them and is phosphorylated by Jak and TyK2 kinases on tyrosine residue Y641 located at the C-terminus of the SH2 domain, as indicated above. This entails the homodimerization of STAT6, which thereby translocates to the nucleus and efficiently binds to DNA sequences through the DBD, acting as a transcription factor ()
[57][75].
Even when at least 12 different
NAB2–STAT6 fusion variants have been described according to their breakpoints
[58][76], the two most recurrent variants are: the
NAB2 exon4–STAT6 exon2 (N4S2), which is the most frequent, and the
NAB2 exon6–STAT6 exon16/17 (N6S16/17). N4S2 entails the fusion between NAB2 that lacks the CID domain and partially lacks the NCD2 domain, and the almost complete STAT6 part. This fusion correlates with a distinctive phenotype: primary tumors are derived mostly from the thoracic cavity, patients are older than in other breakpoint variants, tumors are significantly larger in diameter (median 10 cm) and they exhibit the appearance of a predominantly fibrotic and paucicellular tumor context. In contrast, the N6S16/17 fusion transcript contains almost all the NAB2 portion, except for exon 7 of CID, and a truncated STAT6 protein keeping TAD and part of the C-terminal of the SH2 domain. This N6S16/17 fusion is harbored more commonly by patients with pelvic, meningeal or extremity SFT, younger age than N4S2, smaller tumors (median 4.3 cm) and more tumor cellularity in comparison to N4S2
[58][59][76,77]. The prominent fibrosis seen in N4S2 could be related to the lack of CID (less repression on EGR1), and possibly to the presence of the almost entire STAT6 portion in the chimeric protein. Differential transcriptome analysis between the two most frequent fusion variants revealed that N4S2 exhibited a gene signature enriched for genes involved in DNA binding, gene transcription and nuclear localization, whereas the N6S16/17 signature was enriched for genes involved in tyrosine kinase signaling, cell proliferation and cytoplasmic localization
[60][78]. Despite the fact that N4S2 correlates more frequently with less aggressive SFT, there is no convincing study demonstrating an unequivocal significant prognostic correlation among the fusion variants
[51][69]. Larger studies analyzing homogeneous populations (i.e., completely resected localized tumors, or from metastatic spread) with enough follow-up are required. Additionally, the median tumor size of N4S2 was also greater, which would compensate the apparent better prognosis.