The Ca2+-independent phospholipase A2β (iPLA2β) is a member of the PLA2 family that has been proposed to have roles in multiple biological processes including membrane remodeling, cell proliferation, bone formation, male fertility, cell death, and signaling. Such involvement has led to the identification of iPLA2β activation in several diseases such as cancer, cardiovascular abnormalities, glaucoma, periodontitis, neurological disorders, diabetes, and other metabolic disorders. More recently, there has been heightened interest in the role that iPLA2β plays in promoting inflammation.
- iPLA2β
- macrophages
- T-cells
- inflammation
- eicosanoids
- resolvins
1. Introduction
Phospholipases A2 (PLA2s) hydrolyze the sn-2 substituent of glycerophospholipids to release a lysophospholipid and a free fatty acid
[1]
. Among the family of PLA2s are the group VI Ca
2+
-independent PLA2s (iPLA2s), which include iPLA2β (VIA), iPLA2γ (VIB) iPLA2δ (VIC), iPLA2ε (VID), iPLA2ζ (VIE), and iPLA2η (VIF)
[2]
. The cytosolic iPLA2β enzyme, along with the membrane-associated iPLA2γ, are the most studied of the group VI PLA2s. Because of its emerging link with inflammation, the focus of this review will be on iPLA2β.
Encoded by PLA2G6, the iPLA2β protein (84-88 kDa) has a serine lipase consensus sequence (GTSGT), contains eight N-terminal ankyrin repeats, an ATP binding cassette (GGGVKG), a caspase-3 cleavage site (DVDT), and two calmodulin (CAM) binding domains
. Crystal structure studies suggest that iPLA2β forms a dimer through the interaction of the catalytic domains and that CAM binds to the dimer to cause a closed state, denying the access of substrates to the active site
[4]
. Relief from this inhibitory state is achieved through activation of calmodulin kinase IIβ, which forms a signaling complex with iPLA2β
[5]
. Lipidomics-based LC/MS approaches have revealed that among glycerophospholipids, iPLA2β exhibited the greatest activity towards 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphoethanolamine (PAPE) and, among sn-2 substituents, a greater selectivity for linoleate or myristate
. The currently available selective inhibitors of iPLA2β include the irreversible S-BEL (S-bromoenol lactone) and the reversible 1,1,1-trifluoro-6-(naphthalen-2-yl)hexan-2-one (FKGK18)
. iPLA2β is widely expressed and has many proposed roles, including signal transduction, and is recognized to contribute to neurodegenerative disorders, cancers, myocardial complications, and metabolic dysfunction (reviewed extensively elsewhere
).
While localized in the cytosol under basal conditions
[10]
, iPLA2β mobilizes to subcellular organelles (e.g., the endoplasmic reticulum (ER), mitochondria, nucleus) upon stimulation. Its activation leads to the hydrolysis of the sn-2 fatty acid substituent from membrane glycerophospholipids
to yield a free fatty acid (e.g., arachidonic acid, eicosapentaenoic acid (EPA), docosahexaenoic acid (DHA)) and a 2-lysophospholipid
[17]
. Subsequent metabolism of arachidonic acid by cyclooxygenases (COX), lipoxygenase (LOX), and cytochrome P450 (CYP) pathways leads to the generation of bioactive oxidized eicosanoids, several of which are proinflammatory
[18]
and recognized contributors to inflammatory diseases
[19][20][21][22][23][24][25][26][27]
. Some of the most potent inflammatory eicosanoids
[21]
are prostaglandin E2 (PGE2), leukotrienes (LTs), hydroxyeicosatetraenoic acids (HETEs), and dihydroxyeicosatetraenoic acids (DHETEs), and they contribute to inflammation and autoimmune diseases
[22]
. It is not unexpected that iPLA2β activation can play critical roles in the initiation and progression of inflammatory pathways, which if not curbed can lead to the evolution of a variety of disorders. In contrast, other unsaturated fatty acids such as EPA and DHA can be metabolized to generate pro-resolving lipids, designated as specialized resolving mediators (SPMs)
. Among the proinflammatory lipids generated, PGs and LTs are the first to be produced
[28]
. When there is injury to the tissue, the generated PGs cause pain, swelling, and edema. To resolve the inflammation and clear antigenic debris, SPMs are generated in attempt to restore homeostasis, a process referred to as “lipid mediator class switching”
.
Immune cells express iPLA2β
and iPLA2β-derived lipids (iDLs) contribute to cell proliferation
[37]
, cell cycle progression
[38]
, cell division
[39]
, monocyte migration
[40]
, and superoxide generation
[41]
. Inhibition of iPLA2β reduces reactive oxygen species (ROS) generation
[42]
and is reported to be effective against autoimmune-
[43]
and inflammation-based
diseases.
Although extensive literature exists linking a number of secretory PLA2s (sPLA2s) or cytosolic PLA2α (cPLA2α) with inflammatory responses, only a few studies have described a link between iPLA2β and macrophages, and even fewer have considered a link between iPLA2β and T-cells or B-cells.
2. Protective Consequences of iPLA2
β
Activation
2.1. Cancer Development
Inflammation is a key contributor to cancer development
2.1. Cancer Development
Inflammation is a key contributor to cancer development
[48]
and cytokines released by macrophages and T-cells are integral to this process
[49]
. In view of their earlier observations that iPLA2β-null mice are more susceptible to various inflammatory-based disorders
, Inhoffen et al.
[53]
assessed the ability of immune cells from iPLA2β-null mice to produce cytokines following exposure to CD95/FasL, a trigger of proinflammatory cytokine production
[54]
. They found that iPLA2β-deficiency increased apoptosis in the liver, spleen, and mesenteric lymph nodes (MLN). Although Kupffer cells (i.e., satellite macrophages in the liver) generated a lower production of proinflammatory cytokines TNFα and IL-6, splenocytes became primed to release proinflammatory Th1-/Th17-related cytokines (IFNγ/IL-17α). These findings led to the suggestion that iPLA2β-deficiency can reduce age-related MLN lymphoma development. Mechanistically, they attributed this to the decreased availability of “find-me” and “eat-me” signals derived via iPLA2β activation. These include LPC, a find-me signal
[55]
, which promotes the clearance of apoptotic debris, the accumulation of which triggers and amplifies subsequent immune responses
(Figure 1).
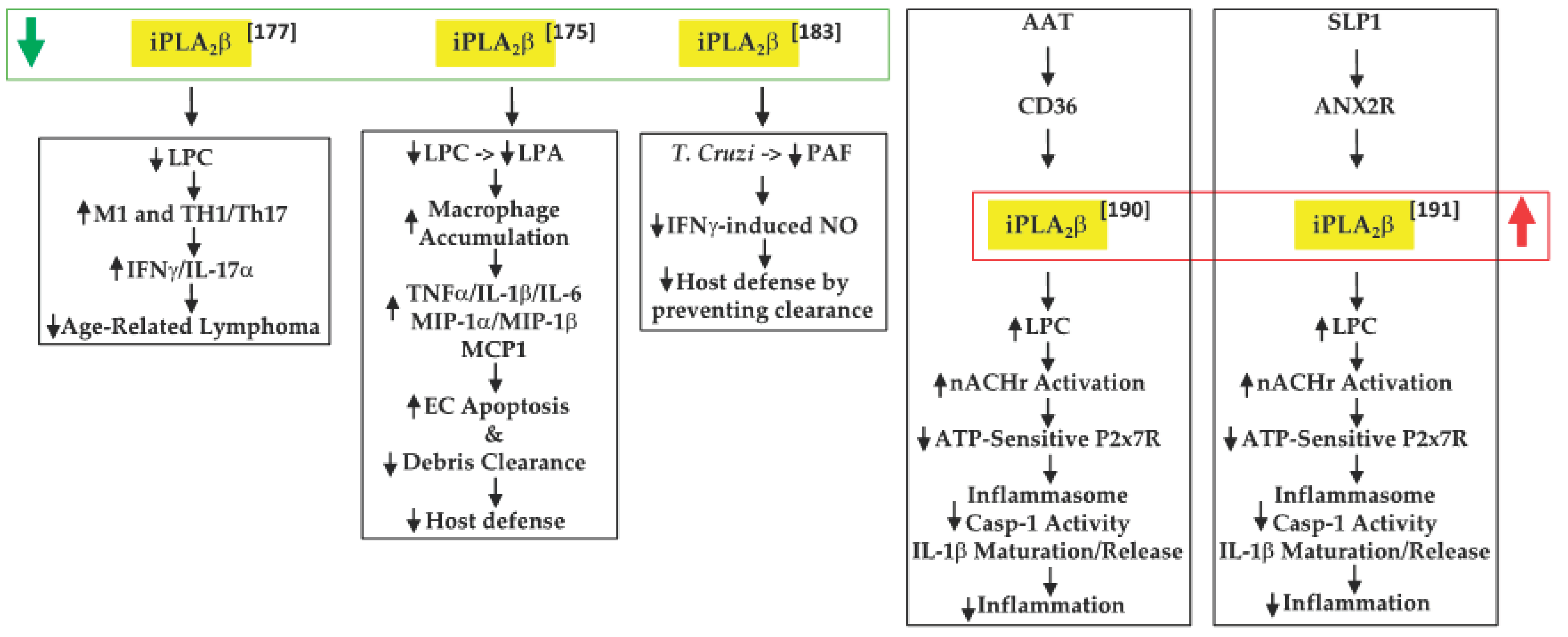
Figure 1. Proposed mechanisms by which iPLA2β inactivation or activation can have protective outcomes in tumor development and promoting host defenses ( green arrow, decreased expression/activity of iPLA2β; red arrow, increased expression/activity of iPLA2β; black up arrow, indicates increase in; black down arrow, indicates decrease in).
Proposed mechanisms by which iPLA2β inactivation or activation can have protective outcomes in tumor development and promoting host defenses ( green arrow, decreased expression/activity of iPLA2β; red arrow, increased expression/activity of iPLA2β; black up arrow, indicates increase in; black down arrow, indicates decrease in).
2.2. Inflammatory Bowel Disease (IBD)
2.2. Inflammatory Bowel Disease (IBD)
An inflammatory disorder such as IBD is a consequence of the dysfunction of the intestinal epithelial barrier and the mucosal immune system
An inflammatory disorder such as IBD is a consequence of the dysfunction of the intestinal epithelial barrier and the mucosal immune system
[58]
. Jiao et al.
[51]
further explored the role of “find-me” signals derived via iPLA2β activation in the context of dextran sodium sulfate-induced IBD. They found that iPLA2β-deficiency promotes the accumulation of infiltrating macrophages and dendritic cells in the colon lamina. These were associated with increased production of inflammatory cytokines (TNFα, IL-1β, and IL-6), macrophage inflammatory proteins (MIP-1α and MIP-1β), and CCL2, leading to intestinal epithelial cell apoptosis and mucus barrier damage. With a concurrent decrease in LPC levels, they speculated that iPLA2β-deficiency mitigated the availability of a “find-me” signal, which limited the clearance of apoptotic debris and the amplification of immune responses. Further, they predicted that the iPLA2β-deficiency also reduced LPA levels, impacting the cellular compass, and preventing the optimal phagocytic function of macrophages. Subsequently, Murase et al.
[59]
, comparing the involvement of various PLA2s, suggested that iPLA2β-deficiency did not worsen the clinical scoring associated with IBD, relative to controls. As they did not reconcile the two studies, it is likely that differences between the source of the iPLA2β
−/− mice and DSS concentrations used may be partly responsible. Furthermore, while a 7-day DSS regimen was employed by both groups, Jiao’s group maintained the mice for an additional 3 days without DSS, prior to analyses. It may also be noted that Murase’s study did demonstrate similar lower clinical scores in the wild-type (WT) and iPLA2β-deficient groups between days 1 and 6, relative to mice with deficiencies in cPLA2α or in a variety of sPLA2s; however, at day 7, the scores in the iPLA2β-deficient group appeared to be as high as in the other PLA2-deficient groups. As such, the role of iPLA2β in this model remains to be clarified.
mice and DSS concentrations used may be partly responsible. Furthermore, while a 7-day DSS regimen was employed by both groups, Jiao’s group maintained the mice for an additional 3 days without DSS, prior to analyses. It may also be noted that Murase’s study did demonstrate similar lower clinical scores in the wild-type (WT) and iPLA2β-deficient groups between days 1 and 6, relative to mice with deficiencies in cPLA2α or in a variety of sPLA2s; however, at day 7, the scores in the iPLA2β-deficient group appeared to be as high as in the other PLA2-deficient groups. As such, the role of iPLA2β in this model remains to be clarified.
2.3. Chagas Disease
2.3. Chagas Disease
A further link between iPLA2β and PAF was reported by McHowat’s group
A further link between iPLA2β and PAF was reported by McHowat’s group
[60]
, in the context of Chagas disease. This disease is caused through infection by the protozoan parasite
[61]
Trypanosoma cruzi (T. cruizi) and can lead to various cardiac abnormalities
[62]
. The sequela of infection begins with induction of an inflammatory response and upregulation of endothelial adhesion molecules
[63], followed by attempts at resolution through the generation of proinflammatory cytokines and the induction of signaling pathways to promote the chemotaxis of immune cells to mitigate the invasion. McHowat’s group reported that infection of iPLA2β-deficient mice resulted in lowered PAF and NO production by cardiac endothelial cells, but that neither the expression of adhesion molecules nor the development of myocardial inflammation was affected. However, significant increases in parasite pseudocysts were noted in the myocardium of iPLA2β-deficient mice. The authors suggested that this was due to an impairment in parasite clearance as a consequence of decreased iPLA2β-mediated LPA production and, as a result, Nox4 expression and NO production. Thus, they surmised that the absence of iPLA2β mitigates parasite clearance due to the reduced recruitment of inflammatory cells to the infected myocardial areas.
, followed by attempts at resolution through the generation of proinflammatory cytokines and the induction of signaling pathways to promote the chemotaxis of immune cells to mitigate the invasion. McHowat’s group reported that infection of iPLA2β-deficient mice resulted in lowered PAF and NO production by cardiac endothelial cells, but that neither the expression of adhesion molecules nor the development of myocardial inflammation was affected. However, significant increases in parasite pseudocysts were noted in the myocardium of iPLA2β-deficient mice. The authors suggested that this was due to an impairment in parasite clearance as a consequence of decreased iPLA2β-mediated LPA production and, as a result, Nox4 expression and NO production. Thus, they surmised that the absence of iPLA2β mitigates parasite clearance due to the reduced recruitment of inflammatory cells to the infected myocardial areas.
2.4. Negative Modulation of Inflammation by AAT and SLP1
2.4. Negative Modulation of Inflammation by AAT and SLP1
In sequential reports, Grau’s group constructed events that participated in regulating IL-1β activation and release. IL-1β is critical to host defense against infections, but the generation of excessive active IL-1β can lead to deleterious inflammatory consequences
In sequential reports, Grau’s group constructed events that participated in regulating IL-1β activation and release. IL-1β is critical to host defense against infections, but the generation of excessive active IL-1β can lead to deleterious inflammatory consequences
[64]
. The release of IL-1β occurs via two signals. The first is an external stimulus that induces the synthesis of pro-IL-1β. The second signal has been suggested to be ATP, which when released from damaged cells activates the purinergic receptor P2 × 7R, promoting the loss of K
+
current and triggering the assembly of NLRP3 (NLR family pyrin domain containing 3)-containing inflammasome
[65]
. This leads to caspase-1 activation, cleavage of pro-IL-1β, and release of IL-1β from the immune cell. Present in inflammatory cells, alpha-1 antitrypsin (AAT) is a strong inducer of anti-inflammatory processes and its upregulation during systemic inflammation has been associated with the decreased production of proinflammatory cytokines, including IL-1β
[66]
. In examining the potential role of AAT in ATP-dependent regulation of IL-1β in their first study
[67]
, Siebers et al. found that AAT signaling through the CD36 receptor activates iPLA2β, which leads to the release of a low molecular weight factor (LMWF). The LMWF is released from the cell and binds to nicotinic acetylcholine receptor (nAchR), leading to inhibition of P2X7R function, prevention of inflammasome assembly, and the processing of pro-IL-1β to active IL-1β, and its release from the cell. These outcomes were significantly mitigated with selective inhibition or knockdown of iPLA2β and were also not evident in PBMCs from iPLA2β-deficient mice. In the second study
[68]
, Zakrzewicz et al. demonstrated that the secretory leukocyte protease inhibitor (SLP1) also interferes with ATP-dependent regulation of IL-1β via iPLA2β activation. SLP1 is also present in inflammatory cells
[69]
and its actions promote anti-inflammatory outcomes. Their results suggested that the SLP1 signals released through annexin 2, a membrane binding protein for SLP1
[70]
, activate iPLA2β, leading to the production of the LMWF and subsequent inhibition of IL-1β maturation and release. Although both pathways were found to mitigate inflammation, unfortunately neither study explored the identity of the LMWF. Interestingly, AAT administration to recent-onset T1D subjects improved β-cell function, which was correlated with reduced IL-1β production from monocytes and myeloid dendritic cells
[71].
.
3. Summary
iPLA2β is a member of the family of PLA2s that hydrolyzes the sn-2 substituent from membrane glycerophospholipids. Thus, activation of iPLA2β can lead to the production of a variety of bioactive lipid mediators. As eicosanoids generated subsequently to iPLA2β-mediated hydrolysis of sn-2 arachidonic acid can exhibit profound proinflammatory effects and SPMs are produced from sn-2 substituents EPA and DHA, the impact of iPLA2β on the inflammatory sequelae is profound. In recent years, the impact of iPLA2β at the immune cell level is being recognized and those studies, as well as those from our laboratory, suggest that iPLA2β activation modulates the maturation, polarization, activation, and functionality of macrophages, T-cells, and B-cells. The continuation of studies addressing these actions of iPLA2β is important and warranted in order to gain a better understanding of the events that lead to the onset, maintenance, and amplification of inflammation. Identifying the relevant selective iDLs with effects on these processes could lead to the development of new strategies to treat autoimmune- and other inflammatory-based diseases.
3. Summary
iPLA2β is a member of the family of PLA2s that hydrolyzes the sn-2 substituent from membrane glycerophospholipids. Thus, activation of iPLA2β can lead to the production of a variety of bioactive lipid mediators. As eicosanoids generated subsequently to iPLA2β-mediated hydrolysis of sn-2 arachidonic acid can exhibit profound proinflammatory effects and SPMs are produced from sn-2 substituents EPA and DHA, the impact of iPLA2β on the inflammatory sequelae is profound. In recent years, the impact of iPLA2β at the immune cell level is being recognized and those studies, as well as those from our laboratory, suggest that iPLA2β activation modulates the maturation, polarization, activation, and functionality of macrophages, T-cells, and B-cells. The continuation of studies addressing these actions of iPLA2β is important and warranted in order to gain a better understanding of the events that lead to the onset, maintenance, and amplification of inflammation. Identifying the relevant selective iDLs with effects on these processes could lead to the development of new strategies to treat autoimmune- and other inflammatory-based diseases.
References
- Gijon, M.A.; Leslie, C.C. Phospholipases A2. Semin. Cell Dev. Biol. 1997, 8, 297–303.
- Turk, J.; White, T.D.; Nelson, A.J.; Lei, X.; Ramanadham, S. iPLA2b and its role in male fertility, neurological disorders, metabolic disorders, and inflammation. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2019, 1864, 846–860.
- Ramanadham, S.; Ali, T.; Ashley, J.W.; Bone, R.N.; Hancock, W.D.; Lei, X. Calcium-independent phospholipases A2 and their roles in biological processes and diseases. J. Lipid Res. 2015, 56, 1643–1668.
- Malley, K.R.; Koroleva, O.; Miller, I.; Sanishvili, R.; Jenkins, C.M.; Gross, R.W.; Korolev, S. The structure of iPLA2beta reveals dimeric active sites and suggests mechanisms of regulation and localization. Nat. Commun. 2018, 9, 765.
- Wang, Z.; Ramanadham, S.; Ma, Z.A.; Bao, S.; Mancuso, D.J.; Gross, R.W.; Turk, J. Group VIA phospholipase A2 forms a signaling complex with the calcium/calmodulin-dependent protein kinase IIbeta expressed in pancreatic islet beta-cells. J. Biol. Chem. 2005, 280, 6840–6849.
- Mouchlis, V.D.; Armando, A.; Dennis, E.A. Substrate-specific inhibition constants for phospholipase A2 acting on unique phospholipid substrates in mixed micelles and membranes using lipidomics. J. Med. Chem. 2019, 62, 1999–2007.
- Mouchlis, V.D.; Chen, Y.; Mccammon, J.A.; Dennis, E.A. Membrane allostery and unique hydrophobic sites promote enzyme substrate specificity. J. Am. Chem. Soc. 2018, 140, 3285–3291.
- Ali, T.; Kokotos, G.; Magrioti, V.; Bone, R.N.; Mobley, J.A.; Hancock, W.; Ramanadham, S. Characterization of FKGK18 as inhibitor of group VIA Ca2+-independent phospholipase A2 (iPLA2b): Candidate drug for preventing beta-cell apoptosis and diabetes. PLoS ONE 2013, 8, e71748.
- Kokotos, G.; Hsu, Y.H.; Burke, J.E.; Baskakis, C.; Kokotos, C.G.; Magrioti, V.; Dennis, E.A. Potent and selective fluoroketone inhibitors of group VIA calcium-independent phospholipase A2. J. Med. Chem. 2010, 53, 3602–3610.
- Gross, R.W.; Ramanadham, S.; Kruszka, K.K.; Han, X.; Turk, J. Rat and human pancreatic islet cells contain a calcium ion independent phospholipase A2 activity selective for hydrolysis of arachidonate which is stimulated by adenosine triphosphate and is specifically localized to islet beta-cells. Biochemistry 1993, 32, 327–336.
- Bao, S.; Jin, C.; Zhang, S.; Turk, J.; Ma, Z.; Ramanadham, S. Beta-cell calcium-independent group VIA phospholipase A2 (iPLA2b): Tracking iPLA2b movements in response to stimulation with insulin secretagogues in INS-1 cells. Diabetes 2004, 53, S186–S189.
- Lei, X.; Zhang, S.; Bohrer, A.; Ramanadham, S. Calcium-independent phospholipase A2 (iPLA2b)-mediated ceramide generation plays a key role in the cross-talk between the endoplasmic reticulum (ER) and mitochondria during ER stress-induced insulin-secreting cell apoptosis. J. Biol. Chem. 2008, 283, 34819–34832.
- Ma, Z.; Ramanadham, S.; Wohltmann, M.; Bohrer, A.; Hsu, F.F.; Turk, J. Studies of insulin secretory responses and of arachidonic acid incorporation into phospholipids of stably transfected insulinoma cells that overexpress group VIA phospholipase A2 (iPLA2b) indicate a signaling rather than a housekeeping role for iPLA2b. J. Biol. Chem. 2001, 276, 13198–13208.
- Ma, Z.; Zhang, S.; Turk, J.; Ramanadham, S. Stimulation of insulin secretion and associated nuclear accumulation of iPLA2b in INS-1 insulinoma cells. Am. J. Physiol. Endocrinol. Metab. 2002, 282, E820–E833.
- Ramanadham, S.; Hsu, F.F.; Zhang, S.; Jin, C.; Bohrer, A.; Song, H.; Bao, S.; Ma, Z.; Turk, J. Apoptosis of insulin-secreting cells induced by endoplasmic reticulum stress is amplified by overexpression of group VIA calcium-independent phospholipase A2 (iPLA2b) and suppressed by inhibition of iPLA2b. Biochemistry 2004, 43, 918–930.
- Song, H.; Bao, S.; Lei, X.; Jin, C.; Zhang, S.; Turk, J.; Ramanadham, S. Evidence for proteolytic processing and stimulated organelle redistribution of iPLA2b. Biochim. Biophys. Acta 2010, 1801, 547–558.
- Ramanadham, S.; Hsu, F.F.; Bohrer, A.; Zhongmin, M.; Turk, J. Studies of the role of group VI phospholipase A2 in fatty acid incorporation, phospholipid remodeling, lysophosphatidylcholine generation, and secretagogue-induced arachidonic acid release in pancreatic islets and insulinoma cells. J. Biol. Chem. 1999, 275, 13915–13927.
- Hanna, V.S.; Hafez, E.A.A. Synopsis of arachidonic acid metabolism: A review. J. Adv. Res. 2018, 11, 23–32.
- Abdulkhaleq, L.A.; Assi, M.A.; Abdullah, R.; Zamri-Saad, M.; Taufiq-Yap, Y.H.; Hezmee, M.N.M. The crucial roles of inflammatory mediators in inflammation: A review. Vet. World 2018, 11, 627–635.
- Khanapure, S.P.; Garvey, D.S.; Janero, D.R.; Letts, L.G. Eicosanoids in inflammation: Biosynthesis, pharmacology, and therapeutic frontiers. Curr. Top. Med. Chem. 2007, 7, 311–340.
- Luo, P.; Wang, M.H. Eicosanoids, beta-cell function, and diabetes. Prostaglandins Other Lipid Mediat. 2011, 95, 1–10.
- Tessaro, F.H.; Ayala, T.S.; Martins, J.O. Lipid mediators are critical in resolving inflammation: A review of the emerging roles of eicosanoids in diabetes mellitus. Biomed. Res. Int. 2015, 2015, 568408.
- Umamaheswaran, S.; Dasari, S.K.; Yang, P.; Lutgendorf, S.K.; Sood, A.K. Stress, inflammation, and eicosanoids: An emerging perspective. Cancer Metastasis Rev. 2018, 37, 203–211.
- Zaitseva, L.; Vaisburd, M.; Shaposhnikova, G.; Mysyakin, E. Role of eicosanoids in regulation of macrophage phagocytic functions by platelet-activating factor during endotoxic shock. Bull. Exp. Biol. Med. 2000, 130, 879–881.
- Kuhn, H.; O’donnell, V.B. Inflammation and immune regulation by 12/15-lipoxygenases. Prog. Lipid Res. 2006, 45, 334–356.
- Issan, Y.; Hochhauser, E.; Guo, A.; Gotlinger, K.H.; Kornowski, R.; Leshem-Lev, D.; Lev, E.; Porat, E.; Snir, E.; Thompson, C.I.; et al. Elevated level of pro-inflammatory eicosanoids and EPC dysfunction in diabetic patients with cardiac ischemia. Prostaglandins Other Lipid Mediat. 2013, 100–101, 15–21.
- Gilroy, D.W.; Newson, J.; Sawmynaden, P.; Willoughby, D.A.; Croxtall, J.D. A novel role for phospholipase A2 isoforms in the checkpoint control of acute inflammation. FASEB J. 2004, 18, 489–498.
- De Jong, A.J.; Kloppenburg, M.; Toes, R.E.; Ioan-Facsinay, A. Fatty acids, lipid mediators, and T-cell function. Front. Immunol. 2014, 5, 483.
- Serhan, C.N. Treating inflammation and infection in the 21st century: New hints from decoding resolution mediators and mechanisms. FASEB J. 2017, 31, 1273–1288.
- Serhan, C.N.; Levy, B.D. Resolvins in inflammation: Emergence of the pro-resolving superfamily of mediators. J. Clin. Investig. 2018, 128, 2657–2669.
- Serhan, C.N. Pro-resolving lipid mediators are leads for resolution physiology. Nature 2014, 510, 92–101.
- Serhan, C.N.; Chiang, N.; Dalli, J.; Levy, B.D. Lipid mediators in the resolution of inflammation. Cold Spring Harb. Perspect. Biol. 2014, 7, a016311.
- Bone, R.N.; Gai, Y.; Magrioti, V.; Kokotou, M.G.; Ali, T.; Lei, X.; Tse, H.M.; Kokotos, G.; Ramanadham, S. Inhibition of Ca2+-independent phospholipase A2beta (iPLA2b) ameliorates islet infiltration and incidence of diabetes in NOD mice. Diabetes 2015, 64, 541–554.
- Gil-De-Gomez, L.; Astudillo, A.M.; Guijas, C.; Magrioti, V.; Kokotos, G.; Balboa, M.A.; Balsinde, J. Cytosolic group IVA and calcium-independent group VIA phospholipase A2s act on distinct phospholipid pools in zymosan-stimulated mouse peritoneal macrophages. J. Immunol. 2014, 192, 752–762.
- Lee, S.H.; Park, D.W.; Park, S.C.; Park, Y.K.; Hong, S.Y.; Kim, J.R.; Lee, C.H.; Baek, S.H. Calcium-independent phospholipase A2beta-Akt signaling is involved in lipopolysaccharide-induced NADPH oxidase 1 expression and foam cell formation. J. Immunol. 2009, 183, 7497–7504.
- Bao, S.; Li, Y.; Lei, X.; Wohltmann, M.; Jin, W.; Bohrer, A.; Semenkovich, C.F.; Ramanadham, S.; Tabas, I.; Turk, J. Attenuated free cholesterol loading-induced apoptosis but preserved phospholipid composition of peritoneal macrophages from mice that do not express group VIA phospholipase A2. J. Biol. Chem. 2007, 282, 27100–27114.
- Ma, Z.; Bohrer, A.; Wohltmann, M.; Ramanadham, S.; Hsu, F.F.; Turk, J. Studies of phospholipid metabolism, proliferation, and secretion of stably transfected insulinoma cells that overexpress group VIA phospholipase A2. Lipids 2001, 36, 689–700.
- Song, Y.; Wilkins, P.; Hu, W.; Murthy, K.S.; Chen, J.; Lee, Z.; Oyesanya, R.; Wu, J.; Barbour, S.E.; Fang, X. Inhibition of calcium-independent phospholipase A2 suppresses proliferation and tumorigenicity of ovarian carcinoma cells. Biochem. J. 2007, 406, 427–436.
- Ashley, J.W.; Hancock, W.D.; Nelson, A.J.; Bone, R.N.; Tse, H.M.; Wohltmann, M.; Turk, J.; Ramanadham, S. Polarization of macrophages toward M2 phenotype is favored by reduction in iPLA2b (Group VIA Phospholipase A2). J. Biol. Chem. 2016, 291, 23268–23281.
- Mishra, R.S.; Carnevale, K.A.; Cathcart, M.K. iPLA2beta: Front and center in human monocyte chemotaxis to MCP-1. J. Exp. Med. 2008, 205, 347–359.
- Ayilavarapu, S.; Kantarci, A.; Fredman, G.; Turkoglu, O.; Omori, K.; Liu, H.; Iwata, T.; Yagi, M.; Hasturk, H.; Van Dyke, T.E. Diabetes-induced oxidative stress is mediated by Ca2+-independent phospholipase A2 in neutrophils. J. Immunol. 2010, 184, 1507–1515.
- Tan, C.; Day, R.; Bao, S.; Turk, J.; Zhao, Q.D. Group VIA phospholipase A2 mediates enhanced macrophage migration in diabetes mellitus by increasing expression of nicotinamide adenine dinucleotide phosphate oxidase 4. Arter. Thromb. Vasc. Biol. 2014, 34, 768–778.
- Kalyvas, A.; Baskakis, C.; Magrioti, V.; Constantinou-Kokotou, V.; Stephens, D.; Lopez-Vales, R.; Lu, J.Q.; Yong, V.W.; Dennis, E.A.; Kokotos, G.; et al. Differing roles for members of the phospholipase A2 superfamily in experimental autoimmune encephalomyelitis. Brain J. Neurol. 2009, 132, 1221–1235.
- Mchowat, J.; Gullickson, G.; Hoover, R.G.; Sharma, J.; Turk, J.; Kornbluth, J. Platelet-activating factor and metastasis: Calcium-independent phospholipase A2beta deficiency protects against breast cancer metastasis to the lung. Am. J. Physiol. Cell Physiol. 2011, 300, C825–C832.
- Nicotera, T.M.; Schuster, D.P.; Bourhim, M.; Chadha, K.; Klaich, G.; Corral, D.A. Regulation of PSA secretion and survival signaling by calcium-independent phopholipase A2beta in prostate cancer cells. Prostate 2009, 69, 1270–1280.
- Scuderi, M.R.; Anfuso, C.D.; Lupo, G.; Motta, C.; Romeo, L.; Guerra, L.; Cappellani, A.; Ragusa, N.; Cantarella, G.; Alberghina, M. Expression of Ca2+-independent and Ca2+-dependent phospholipases A2 and cyclooxygenases in human melanocytes and malignant melanoma cell lines. Biochim. Biophys. Acta 2008, 1781, 635–642.
- Thayer, T.C.; Delano, M.; Liu, C.; Chen, J.; Padgett, L.E.; Tse, H.M.; Annamali, M.; Piganelli, J.D.; Moldawer, L.L.; Mathews, C.E. Superoxide production by macrophages and T cells is critical for the induction of autoreactivity and type 1 diabetes. Diabetes 2011, 60, 2144–2151.
- Aggarwal, B.B.; Vijayalekshmi, R.V.; Sung, B. Targeting inflammatory pathways for prevention and therapy of cancer: Short-term friend, long-term foe. Clin. Cancer Res. 2009, 15, 425–430.
- Lin, W.W.; Karin, M. A cytokine-mediated link between innate immunity, inflammation, and cancer. J. Clin. Investig. 2007, 117, 1175–1183.
- Jiao, L.; Gan-Schreier, H.; Tuma-Kellner, S.; Stremmel, W.; Chamulitrat, W. Sensitization to autoimmune hepatitis in group VIA calcium-independent phospholipase A2-null mice led to duodenal villous atrophy with apoptosis, goblet cell hyperplasia and leaked bile acids. Biochim. Biophys. Acta 2015, 1852, 1646–1657.
- Jiao, L.; Inhoffen, J.; Gan-Schreier, H.; Tuma-Kellner, S.; Stremmel, W.; Sun, Z.; Chamulitrat, W. Deficiency of group VIA phospholipase A2 (iPLA2b) renders susceptibility for chemical-induced colitis. Dig. Dis. Sci. 2015, 60, 3590–3602.
- Xu, W.; Tuma, S.; Katava, N.; Pathil-Warth, A.; Stremmel, W.; Chamultrat, W. Deficiencies of calcium-independent phospholipase A2 B in vivo causes reducedsystemic lipids and lipoproteins concomitant with increased hepatic apoptosis and inflammation. In Journal of Hepathology, Proceeding of The International Liver CongressTM (EASL), Vienna, Austria, 10–14 April 2012; EASL: Barcelona, Spain, 2012.
- Inhoffen, J.; Tuma-Kellner, S.; Straub, B.; Stremmel, W.; Chamulitrat, W. Deficiency of iPLA2beta primes immune cells for proinflammation: Potential involvement in age-related mesenteric lymph node lymphoma. Cancers 2015, 7, 2427–2442.
- Park, D.R.; Thomsen, A.R.; Frevert, C.W.; Pham, U.; Skerrett, S.J.; Kiener, P.A.; Liles, W.C. Fas (CD95) induces proinflammatory cytokine responses by human monocytes and monocyte-derived macrophages. J. Immunol. 2003, 170, 6209–6216.
- Lauber, K.; Bohn, E.; Krober, S.M.; Xiao, Y.J.; Blumenthal, S.G.; Lindemann, R.K.; Marini, P.; Wiedig, C.; Zobywalski, A.; Baksh, S.; et al. Apoptotic cells induce migration of phagocytes via caspase-3-mediated release of a lipid attraction signal. Cell 2003, 113, 717–730.
- Albert, M.L. Death-defying immunity: Do apoptotic cells influence antigen processing and presentation? Nat. Rev. Immunol. 2004, 4, 223–231.
- Scaffidi, P.; Misteli, T.; Bianchi, M.E. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature 2002, 418, 191–195.
- Xavier, R.J.; Podolsky, D.K. Unravelling the pathogenesis of inflammatory bowel disease. Nature 2007, 448, 427–434.
- Murase, R.; Sato, H.; Yamamoto, K.; Ushida, A.; Nishito, Y.; Ikeda, K.; Kobayashi, T.; Yamamoto, T.; Taketomi, Y.; Murakami, M. Group X secreted phospholipase A2 releases omega3 polyunsaturated fatty acids, suppresses colitis, and promotes sperm fertility. J. Biol. Chem. 2016, 291, 6895–6911.
- Sharma, J.; Blase, J.R.; Hoft, D.F.; Marentette, J.O.; Turk, J.; Mchowat, J. Mice with genetic deletion of group via phospholipase a2beta exhibit impaired macrophage function and increased parasite load in Trypanosoma cruzi-induced myocarditis. Infect. Immun. 2016, 84, 1137–1142.
- Bern, C.; Verastegui, M.; Gilman, R.H.; Lafuente, C.; Galdos-Cardenas, G.; Calderon, M.; Pacori, J.; Del Carmen Abastoflor, M.; Aparicio, H.; Brady, M.F.; et al. Congenital Trypanosoma cruzi transmission in Santa Cruz, Bolivia. Clin. Infect. Dis. 2009, 49, 1667–1674.
- Rassi, A., Jr.; Rassi, A.; Little, W.C. Chagas’ heart disease. Clin. Cardiol. 2000, 23, 883–889.
- Lannes-Vieira, J.; Silverio, J.C.; Pereira, I.R.; Vinagre, N.F.; Carvalho, C.M.; Paiva, C.N.; Silva Da, A.A. Chronic Trypanosoma cruzi-elicited cardiomyopathy: From the discovery to the proposal of rational therapeutic interventions targeting cell adhesion molecules and chemokine receptors–How to make a dream come true. Mem. Inst. Oswaldo Cruz 2009, 104, 226–235.
- Dinarello, C.A.; Simon, A.; Van Der Meer, J.W. Treating inflammation by blocking interleukin-1 in a broad spectrum of diseases. Nat. Rev. Drug Discov. 2012, 11, 633–652.
- Lamkanfi, M.; Dixit, V.M. Mechanisms and functions of inflammasomes. Cell 2014, 157, 1013–1022.
- Ehlers, M.R. Immune-modulating effects of alpha-1 antitrypsin. Biol. Chem. 2014, 395, 1187–1193.
- Siebers, K.; Fink, B.; Zakrzewicz, A.; Agne, A.; Richter, K.; Konzok, S.; Hecker, A.; Zukunft, S.; Kullmar, M.; Klein, J.; et al. Alpha-1 Antitrypsin Inhibits ATP-Mediated Release of Interleukin-1beta via CD36 and Teceptors. Front. Immunol. 2018, 9, 877.
- Zakrzewicz, A.; Richter, K.; Zakrzewicz, D.; Siebers, K.; Damm, J.; Agne, A.; Hecker, A.; Mcintosh, J.M.; Chamulitrat, W.; Krasteva-Christ, G.; et al. SLPI Inhibits ATP-mediated maturation of IL-1beta in human monocytic leukocytes: A Tyer. Front. Immunol. 2019, 10, 664.
- Moreau, T.; Baranger, K.; Dade, S.; Dallet-Choisy, S.; Guyot, N.; Zani, M.L. Multifaceted roles of human elafin and secretory leukocyte proteinase inhibitor (SLPI), two serine protease inhibitors of the chelonianin family. Biochimie 2008, 90, 284–295.
- Ma, G.; Greenwell-Wild, T.; Lei, K.; Jin, W.; Swisher, J.; Hardegen, N.; Wild, C.T.; Wahl, S.M. Secretory leukocyte protease inhibitor binds to annexin II, a cofactor for macrophage HIV-1 infection. J. Exp. Med. 2004, 200, 1337–1346.
- Gottlieb, P.A.; Alkanani, A.K.; Michels, A.W.; Lewis, E.C.; Shapiro, L.; Dinarello, C.A.; Zipris, D. alpha1-Antitrypsin therapy downregulates toll-like receptor-induced IL-1beta responses in monocytes and myeloid dendritic cells and may improve islet function in recently diagnosed patients with type 1 diabetes. J. Clin. Endocrinol. Metab. 2014, 99, E1418–E1426.