Antigenic essence – the part of a cell that is both available to the immune system and also highly specific to cell type on a molecular profile level. Antigenic essence can be collected from the cell surface by treating living cells with protease (trypsin) under mild conditions. Cells are a natural source for the entire diversity of native antigens including for anticancer vaccination. Antigenic essence takes advantage of this while also minimizing the limitations associated with the use of whole cells for anticancer vaccination.
- antigenic essence
- cancer vaccine
- proteomic footprint
- mass spectrometry
1. Introduction
2. Antigenic Essence Concept
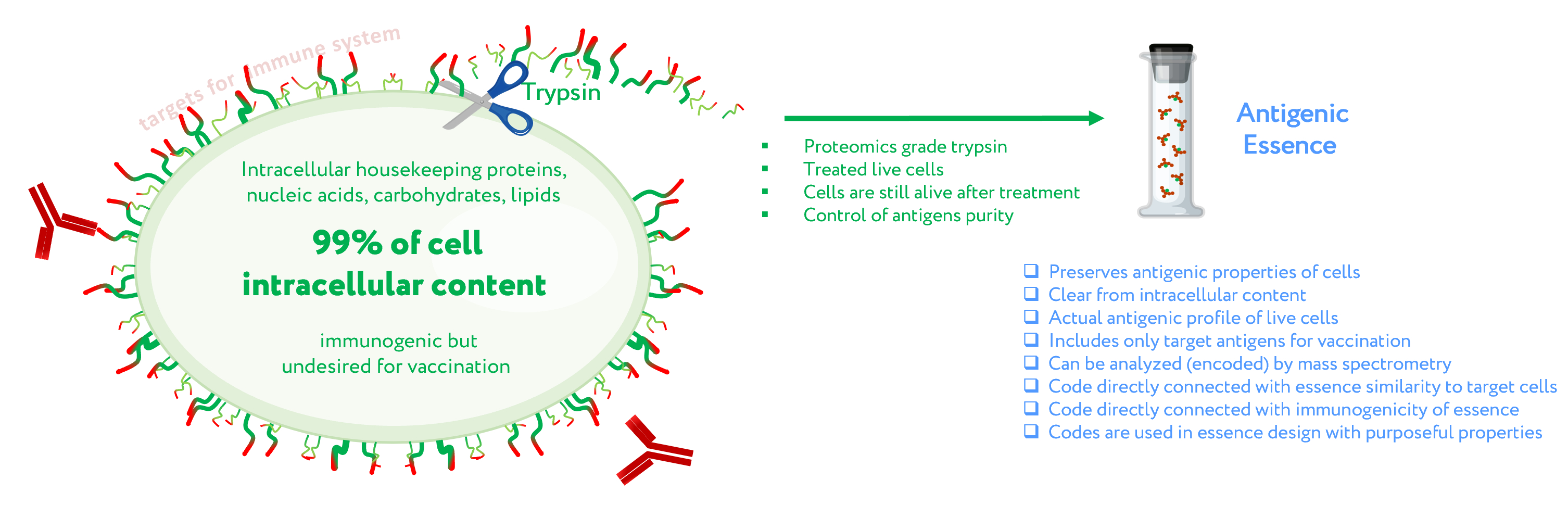
3. Antigenic Essence Equivalence to Cell Antigens
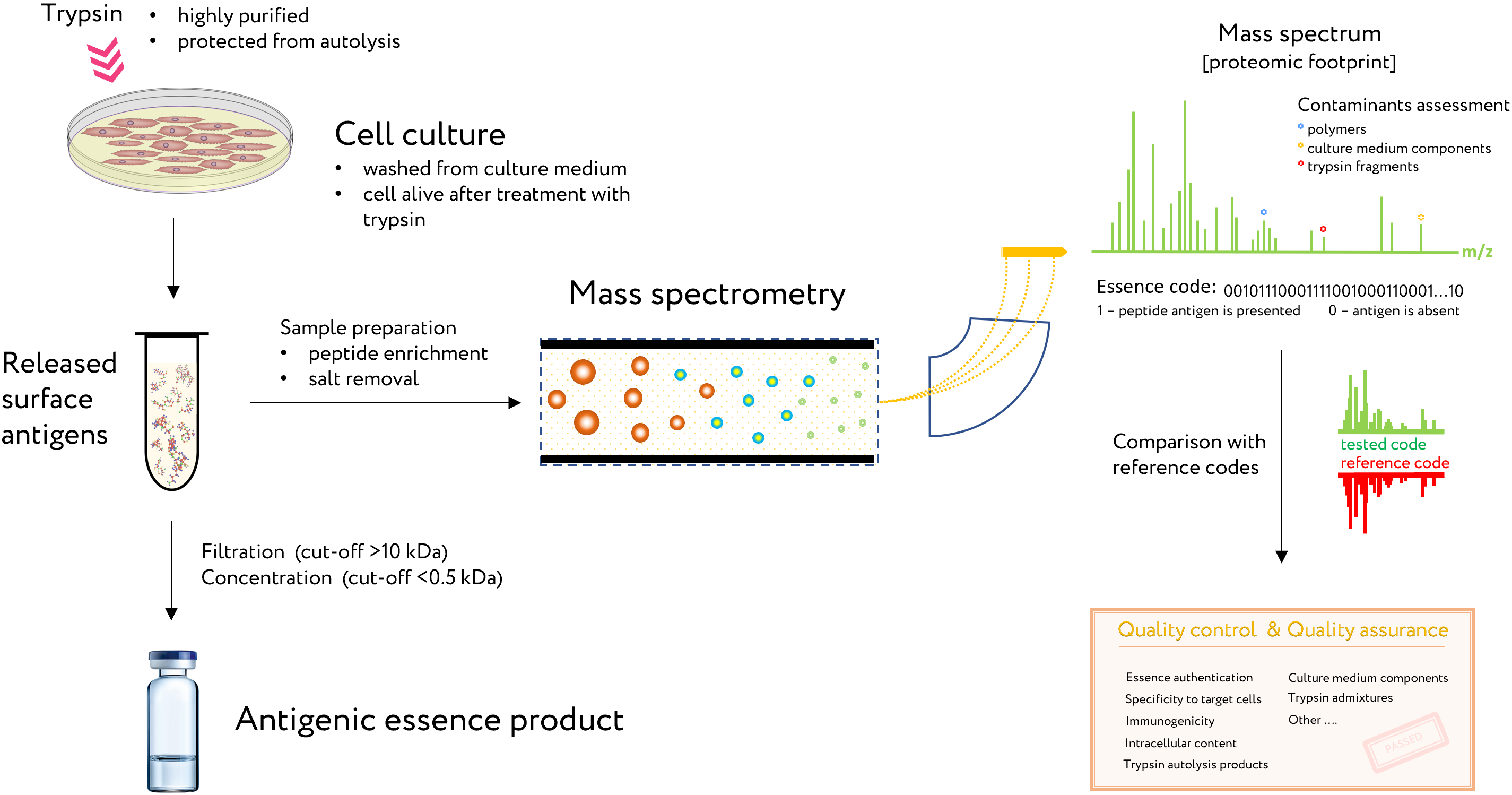
4. Control of Antigenic Essence Composition
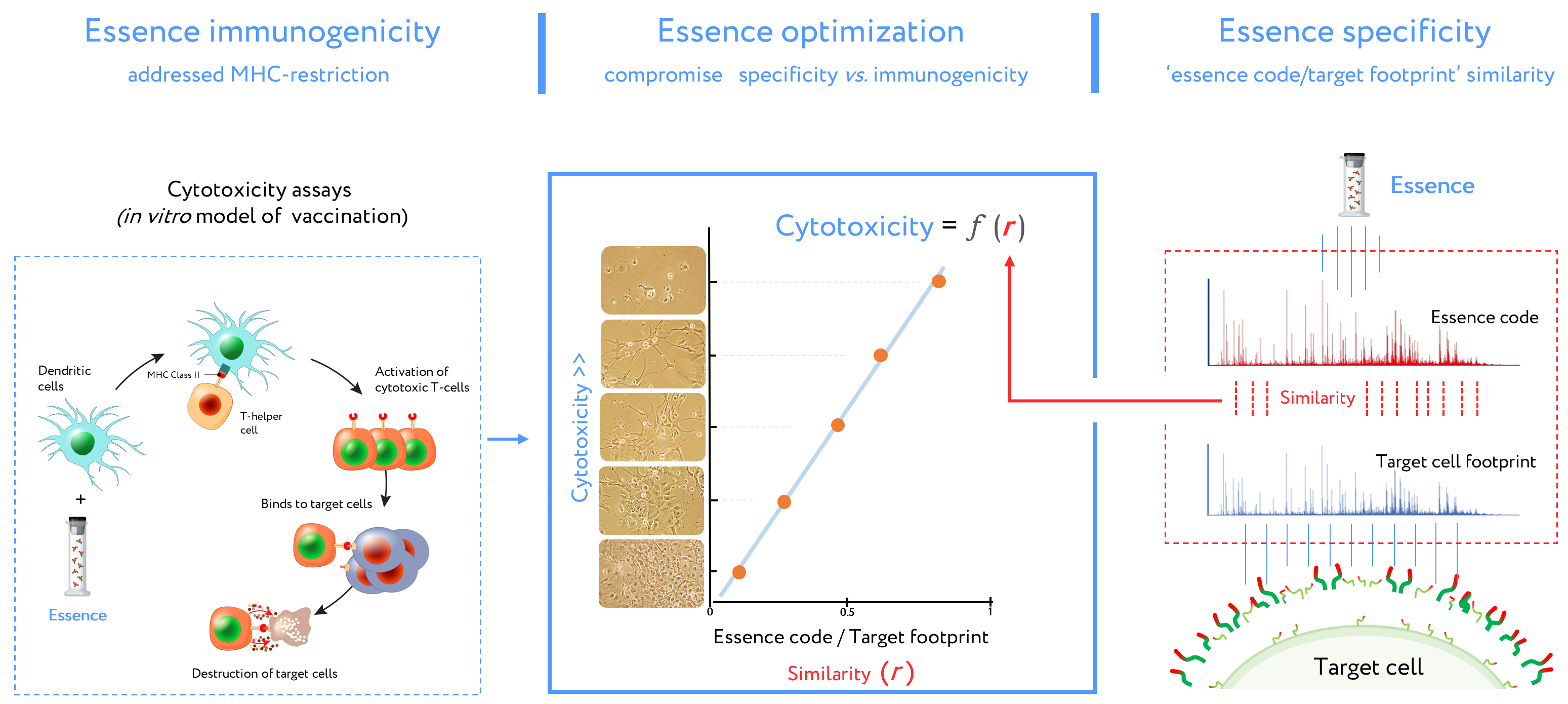
5. Antigens Size in Essence and MHC Restriction
6. Antigenic Essence Composition and MHC Mediated Immunogenicity
7. Antigenic Essence and Immunopeptidome
8. Upgrade of Developed Cellular Cancer Vaccines
Developed to date classical cancer vaccines, which operate on the principle that you should vaccinate with what you want to develop protection against, include whole-cell formulations. Such vaccines have been widely tested in various cancers and their advantages are described but such vaccines have not passed clinical trials due to lack of effectiveness. Antigenic essence technology offers the possibility to revitalize the field of whole-cell-based vaccination. In such vaccines, whole cells just may be replaced by antigenic essence obtained from the same cancer cells.
In addition to all the advantages described above that antigenic essence gives during vaccination, some other advantages of such upgrade of vaccines should be mentioned. Since antigenic essence is acellular formulation, such upgraded vaccines will have undeniable advantages concerning safety. They are free of any cells and supramolecular formations. The production process excludes the presence of bacteria, viruses, protozoa, as well as prions and other foreign proteins (e.g., bovine proteins from the culture medium). Thus, they are less likely to cause infection or allergic reactions. This aligns with current guidance regarding the development of new cancer vaccines with enhanced safety and tolerability profiles [55] and simplifies the implementation of Food and Drug Administration (FDA) requirements [56]. The absence of side effects will significantly reduce the door-to-needle time, which is extremely important for mass vaccination and has been a significant limiting and price-forming factor of cellular vaccines.
References
- M. G. Hanna; L. C. Peters; Specific immunotherapy of established visceral micrometastases by BCG-tumor cell vaccine alone or as an adjunct to surgery. Cancer 1978, 42, 2613-2625.
- Milton Berger; Fernando T Kreutz; Jorge L Horst; Aline C Baldi; Walter J Koff; Phase I study with an autologous tumor cell vaccine for locally advanced or metastatic prostate cancer. Journal of Pharmacy & Pharmaceutical Sciences 2007, 10, 144–152.
- Jules E. Harris; Louise Ryan; Herbert C. Hoover Jr; Robert K. Stuart; Martin M. Oken; Al B. Benson; Edward Mansour; Daniel G. Haller; Judith Manola; Michael G. Hanna Jr; et al. Adjuvant Active Specific Immunotherapy for Stage II and III Colon Cancer With an Autologous Tumor Cell Vaccine: Eastern Cooperative Oncology Group Study E5283. Journal of Clinical Oncology 2000, 18, 148-148.
- C Maver; M McKneally; Preparation of autologous tumor cell vaccine from human lung cancer. Cancer Research 1979, 39, 3276.
- R S Schulof; D Mai; M A Nelson; H M Paxton; J W Cox; M L Turner; M Mills; W R Hix; L E Nochomovitz; L C Peters; et al. Active specific immunotherapy with an autologous tumor cell vaccine in patients with resected non-small cell lung cancer. Molecular biotherapy 1988, 1, 30–36.
- John Nemunaitis; Daniel Sterman; David Jablons; John W. Smith; Bernard Fox; Phil Maples; Scott Hamilton; Flavia Borellini; Andy Lin; Sayeh Morali; et al.Kristen Hege Granulocyte-Macrophage Colony-Stimulating Factor Gene-Modified Autologous Tumor Vaccines in Non-Small-Cell Lung Cancer. Journal of the National Cancer Institute 2004, 96, 326-331.
- Dominik Rüttinger; Natasja K Van Den Engel; Hauke Winter; Marcus Schlemmer; Heike Pohla; Stefanie Grützner; Beate Wagner; Dolores J Schendel; Bernard A Fox; K-W Jauch; et al.Rudolf A Hatz Adjuvant therapeutic vaccination in patients with non-small cell lung cancer made lymphopenic and reconstituted with autologous PBMC: first clinical experience and evidence of an immune response. Journal of Translational Medicine 2007, 5, 43.
- Vincent A. De Weger; Annelies W. Turksma; Quirinus J.M. Voorham; Zelda Euler; Herman Bril; Alfons J. Van Den Eertwegh; Elisabeth Bloemena; Herbert M. Pinedo; Jan B. Vermorken; Harm Van Tinteren; et al.Gerrit A. MeijerErik Hooijberg Clinical Effects of Adjuvant Active Specific Immunotherapy Differ between Patients with Microsatellite-Stable and Microsatellite-Instable Colon Cancer. Clinical Cancer Research 2012, 18, 882-889.
- Michael G. Hanna; Herbert C. Hoover; Jan B. Vermorken; Jules E. Harris; Herbert M. Pinedo; Adjuvant active specific immunotherapy of stage II and stage III colon cancer with an autologous tumor cell vaccine: first randomized phase III trials show promise. Vaccine 2001, 19, 2576-2582.
- D Ockert; V Schirrmacher; N Beck; E Stoelben; T Ahlert; J Flechtenmacher; E Hagmüller; R Buchcik; M Nagel; H D Saeger; et al. Newcastle disease virus-infected intact autologous tumor cell vaccine for adjuvant active specific immunotherapy of resected colorectal carcinoma.. Clinical Cancer Research 1996, 2, 21–28.
- A Baars; J M G H Van Riel; M A Cuesta; E H Jaspars; H M Pinedo; A J M Van Den Eertwegh; Metastasectomy and active specific immunotherapy for a large single melanoma metastasis. Hepatogastroenterology 2002, 49, 691–693.
- D Berd; H C Maguire; P McCue; M J Mastrangelo; Treatment of metastatic melanoma with an autologous tumor-cell vaccine: clinical and immunologic results in 64 patients.. Journal of Clinical Oncology 1990, 8, 1858-1867.
- Rosa Méndez; Francisco Ruiz-Cabello; Teresa Rodríguez; Ana Del Campo; Annette Paschen; Dirk Schadendorf; Federico Garrido; Identification of different tumor escape mechanisms in several metastases from a melanoma patient undergoing immunotherapy. Cancer Immunology, Immunotherapy 2007, 56, 88-94.
- Scott J Antonia; John Seigne; Jose Diaz; Carlos Muro-Cacho; Martine Extermann; Mary Jane Farmelo; Maria Friberg; Marwan Alsarraj; J J Mahany; Julio Pow-Sang; et al.Alan CantorWilliam Janssen Phase I trial of a B7-1 (CD80) gene modified autologous tumor cell vaccine in combination with systemic interleukin-2 in patients with metastatic renal cell carcinoma. Journal of Urology 2002, 167, 1995–2000.
- Mayer Fishman; Terri B. Hunter; Hatem Soliman; Patricia Thompson; Mary Dunn; Renee Smilee; Mary Jane Farmelo; David R. Noyes; John J. Mahany; Ji-Hyun Lee; et al.Alan CantorJane MessinaJohn SeigneJulio Pow-SangWilliam JanssenScott J. Antonia Phase II Trial of B7-1 (CD-86) Transduced, Cultured Autologous Tumor Cell Vaccine Plus Subcutaneous Interleukin-2 for Treatment of Stage IV Renal Cell Carcinoma. Journal of Immunotherapy 2008, 31, 72-80.
- Y Kinoshita; T Kono; R Yasumoto; T Kishimoto; C Y Wang; G P Haas; N Nishisaka; Antitumor effect on murine renal cell carcinoma by autologous tumor vaccines genetically modified with granulocyte-macrophage colony-stimulating factor and interleukin-6 cells.. Journal of Immunotherapy 2001, 24, 205–211.
- Petr G. Lokhov; Elena E. Balashova; Cellular Cancer Vaccines: an Update on the Development of Vaccines Generated from Cell Surface Antigens. Journal of Cancer 2010, 1, 230-241.
- Elena E Balashova; Petr G. Lokhov; Proteolytically-cleaved Fragments of Cell-surface Proteins from Live Tumor Cells Stimulate Anti-tumor Immune Response In vitro. Journal of Carcinogenesis & Mutagenesis 2010, 1, 1-3.
- Elena E. Balashova; Petr G. Lokhov; Proteolytically-cleaved Fragments of Cell Surface Proteins Stimulate a Cytotoxic Immune Response Against Tumoractivated Endothelial Cells In vitro. Journal of Cancer Science & Therapy 2010, 2, 126-131.
- Lokhov, P.G. Method for Producing An Antitumoral Vaccine Based on Surface Endothelial Cell Antigens. U.S. Patent Application No. 9844586, 19 December 2007
- G. N. Stacey; Cell contamination leads to inaccurate data: we must take action now. Nature 2000, 403, 356-356.
- Roderick A.F. MacLeod; Wilhelm G. Dirks; Yoshinobu Matsuo; Maren Kaufmann; Herbert Milch; Hans G. Drexler; Widespread intraspecies cross-contamination of human tumor cell lines arising at source. International Journal of Cancer 1999, 83, 555-563.
- Editoral.; Identity crisis. Nature 2009, 457, 935-936.
- C. M. Cabrera; F. Cobo; A. Nieto; J. L. Cortes; R. M. Montes; Purificación Catalina; A. Concha; Identity tests: determination of cell line cross-contamination. Cytotechnology 2006, 51, 45-50.
- L. Hayflick; P.S. Moorhead; The serial cultivation of human diploid cell strains. Experimental Cell Research 1961, 25, 585-621.
- Woodring E. Wright; Jerry W. Shay; Historical claims and current interpretations of replicative aging. Nature Biotechnology 2002, 20, 682-688.
- John W. Huggins; Robert W. Chestnut; Norman N. Durham; Kermit L. Carraway; Molecular changes in cell surface membranes resulting from trypsinization of sarcoma 180 tumor cells. Biochimica et Biophysica Acta (BBA) - Biomembranes 1976, 426, 630-637.
- Rachel Angus; C.M.P. Collins; M.O. Symes; Expression of major histocompatibility complex (MHC) antigens and their loss on culture in renal carcinoma. European Journal of Cancer 1993, 29, 2158-2160.
- Petr Lokhov; Elena Balashova; Maxim Dashtiev; Cell proteomic footprint. Rapid Communications in Mass Spectrometry 2009, 23, 680-682.
- Nabholz, M.; MacDonald, H.R.; Cytolytic T Lymphocytes. Annu. Rev. Immunol. 1983, 1, 273.
- A Lanzavecchia; Identifying strategies for immune intervention. Science 1993, 260, 937-944.
- Gideon Berke; The Binding and Lysis of Target Cells by Cytotoxic Lymphocytes: Molecular and Cellular Aspects. Annual Review of Immunology 1994, 12, 735-773.
- Ryan J. Malonis; Jonathan R. Lai; Olivia Vergnolle; Peptide-Based Vaccines: Current Progress and Future Challenges. Chemical Reviews 2019, 120, 3210-3229.
- B J Van Den Eynde; T Boon; Tumor antigens recognized by T lymphocytes. International Journal of Clinical & Laboratory Research 1997, 27, 267–268.
- Steven A. Rosenberg; Cancer vaccines based on the identification of genes encoding cancer regression antigens. Immunology Today 1997, 18, 175-182.
- B Fisk; T L Blevins; J T Wharton; C G Ioannides; Identification of an immunodominant peptide of HER-2/neu protooncogene recognized by ovarian tumor-specific cytotoxic T lymphocyte lines.. Journal of Experimental Medicine 1995, 181, 2109-2117.
- G. E. Peoples; P. S. Goedegebuure; R. Smith; D. C. Linehan; I. Yoshino; T. J. Eberlein; Breast and ovarian cancer-specific cytotoxic T lymphocytes recognize the same HER2/neu-derived peptide.. Proceedings of the National Academy of Sciences 1995, 92, 432-436.
- A. Cox; J Skipper; Y Chen; R. Henderson; T. Darrow; J Shabanowitz; V. Engelhard; D. Hunt; C. Slingluff; Identification of a peptide recognized by five melanoma-specific human cytotoxic T cell lines. Science 1994, 264, 716-719.
- Pierre G. Coulie; Benoît J. Van Den Eynde; Pierre Van Der Bruggen; Thierry Boon-Falleur; Tumour antigens recognized by T lymphocytes: at the core of cancer immunotherapy. Nature Cancer 2014, 14, 135-146.
- E Celis; A Sette; H M Grey; Epitope selection and development of peptide based vaccines to treat cancer.. Seminars in Cancer Biology 1995, 6, 329.
- E Appella; D J Loftus; K Sakaguchi; A Sette; E Celis; Synthetic antigenic peptides as a new strategy for immunotherapy of cancer. Biomedical peptides, proteins & nucleic acids : structure, synthesis & biological activity 1995, 1, 177.
- Esteban Celis; John Fikes; Peggy Wentworth; John Sidney; Scott Southwood; Ajesh Maewal; Marie-France Del Guercio; Alessandro Sette; Brian Livingston; Identification of potential CTL epitopes of tumor-associated antigen MAGE-1 for five common HLA-A alleles. Molecular Immunology 1994, 31, 1423-1430.
- V Tsai; S Southwood; J Sidney; Kazuyasu Sakaguchi; Y Kawakami; E Appella; A Sette; E Celis; Identification of subdominant CTL epitopes of the GP100 melanoma-associated tumor antigen by primary in vitro immunization with peptide-pulsed dendritic cells. The Journal of Immunology 1997, 158, 1796.
- E. Celis; V. Tsai; C. Crimi; R. DeMars; P. A. Wentworth; R. W. Chesnut; H. M. Grey; A. Sette; H. M. Serra; Induction of anti-tumor cytotoxic T lymphocytes in normal humans using primary cultures and synthetic peptide epitopes.. Proceedings of the National Academy of Sciences 1994, 91, 2105-2109.
- P Weynants; B Lethé; F Brasseur; M Marchand; T Boon; Expression of mage genes by non-small-cell lung carcinomas. International Journal of Cancer 1994, 56, 826.
- R. G. Vincent; T. M. Chu; T. B. Fergen; M. Ostrander; Carcinoembryonic antigen in 228 patients with carcinoma of the lung. Cancer 2010, 36, 2069-2076.
- D J Slamon; W Godolphin; L A Jones; J A Holt; S G Wong; D E Keith; W J Levin; S G Stuart; J Udove; A Ullrich; et al.Al. Et Studies of the HER-2/neu proto-oncogene in human breast and ovarian cancer. Science 1989, 244, 707-712.
- Ichiro Kawashima; Stephen J Hudson; Van Tsai; Scott Southwood; Kazutoh Takesako; Ettore Appella; Alessandro Sette; Esteban Celis; The Multi-epitope Approach for Immunotherapy for Cancer: Identification of Several CTL Epitopes from Various Tumor-Associated Antigens Expressed on Solid Epithelial Tumors. Human Immunology 1998, 59, 1-14.
- Shunji Sugawara; Toru Abo; Katsuo Kumagai; A simple method to eliminate the antigenicity of surface class I MHC molecules from the membrane of viable cells by acid treatment at pH 3. Journal of Immunological Methods 1987, 100, 83-90.
- M. Bassani-Sternberg; E. Barnea; I. Beer; I. Avivi; T. Katz; A. Admon; Soluble plasma HLA peptidome as a potential source for cancer biomarkers. Proceedings of the National Academy of Sciences 2010, 107, 18769-18776.
- S. Demaria; R. Schwab; S.R. Gottesman; Y. Bushkin; Soluble beta 2-microglobulin-free class I heavy chains are released from the surface of activated and leukemia cells by a metalloprotease.. Journal of Biological Chemistry 1994, 269, 6689-6694.
- Sandra Demaria; Yuri Bushkin; Soluble HLA proteins with bound peptides are released from the cell surface by the membrane metalloproteinase. Human Immunology 2000, 61, 1332-1338.
- Giacomo Galati; Cinzia Arcelloni; Rita Paroni; Silvia Heltai; Patrizia Rovere; Claudio Rugarli; Angelo A. Manfredi; Quantitative cytometry of MHC class I digestion from living cells. Cytometry 1997, 27, 77-83.
- Kwasi Antwi; Paul D. Hanavan; Cheryl E. Myers; Yvette W. Ruiz; Eric J. Thompson; Douglas F. Lake; Proteomic identification of an MHC-binding peptidome from pancreas and breast cancer cell lines. Molecular Immunology 2009, 46, 2931-2937.
- Alex Kudrin; Overview of cancer vaccines. Human Vaccines & Immunotherapeutics 2012, 8, 1335-1353.
- [Experts]; Guidance for Industry Clinical Considerations for Therapeutic Cancer Vaccines. Biotechnology Law Report 2012, 31, 303-309.
- Andrew N. Hoofnagle; Jeffrey R. Whiteaker; Steven A. Carr; Eric Kuhn; Tao Liu; Sam A. Massoni; Stefani N. Thomas; Raymond R Townsend; Lisa J. Zimmerman; Emily S Boja; et al.Jing ChenDaniel L. CrimminsSherri DaviesYuqian GaoTara R. HiltkeKaren A. KetchumChristopher R. KinsingerMehdi MesriMatthew R. MeyerWei-Jun QianRegine M. SchoenherrMitchell G. ScottTujin ShiGordon R. WhiteleyJohn A. WrobelChaochao WuBrad L. AckermannRuedi AebersoldDavid R. BarnidgeDavid M. BunkNigel J ClarkeJordan B. FishmanRussell P GrantUlrike KusebauchMark M. KushnirMark S. LowenthalRobert L. MoritzHendrik NeubertScott D. PattersonAlan L. RockwoodJohn Charles RogersRavinder J. SinghJennifer E Van EykSteven H WongShucha ZhangDaniel W. ChanXian ChenMatthew J. EllisDaniel LieblerKarin D. RodlandHenry RodriguezRichard D. SmithZhen ZhangHui ZhangAmanda G. Paulovich Recommendations for the Generation, Quantification, Storage, and Handling of Peptides Used for Mass Spectrometry–Based Assays. Clinical Chemistry 2016, 62, 48-69.
- Hsiang-Ling Huang; Hsiang-Wei Hsing; Tzu-Chia Lai; Yi-Wen Chen; Tian-Ren Lee; Hsin-Tsu Chan; Ping-Chiang Lyu; Chieh-Lin Wu; Ying-Chieh Lu; Szu-Ting Lin; et al.Cheng-Wen LinChih-Ho LaiHao-Teng ChangHsiu-Chuan ChouHong-Lin Chan Trypsin-induced proteome alteration during cell subculture in mammalian cells. Journal of Biomedical Science 2010, 17, 36-36.