Type III Secretion Systems (T3SSs) are multicomponent nanomachines located at the cell envelope of Gram-negative bacteria. Their main function is to transport bacterial proteins either extracellularly or directly into the eukaryotic host cell cytoplasm. Type III Secretion effectors (T3SEs), latest to be secreted T3S substrates, are destined to act at the eukaryotic host cell cytoplasm and occasionally at the nucleus, hijacking cellular processes through mimicking eukaryotic proteins. T3SE families adopt novel folds to target eukaryotic functions. These folds comprise a high helical content, which possibly reflects the specific requirements from T3SS effectors. In particular, effectors must (i) be able to be easily unfolded, (ii) cross the narrow T3S channel, (iii) be highly folded as soon as they will be found inside the host cell, in order to evade the host defense mechanisms, and (iv) display functional competence and structural plasticity in their final destination. α-helices can optimally fulfil these requirements.
- Type III Secretion System (T3SS)
- Type III Secretion effector (T3SE)
- dictionary of secondary structure in proteins (DSSP)
- α-helical domains
1. Type III Secretion Systems
Type III Secretion Systems (T3SSs) are multiprotein nanomachines crossing the three main physical barriers of Gram-negative bacteria: the inner cell membrane, the peptidoglycan layer and the outer bacterial membrane. These systems, originating from the bacterial flagellum, have further evolved and diversified to bypass additional physical barriers: the host cell membrane and, in the case of plant pathogens, the cell wall [1][2][3]. Their ultimate task is to deliver bacterial effector proteins (T3SEs) to the host cell cytoplasm in order to hijack the eukaryotic cell metabolism.
The secretion core of these nanomachines and the secretion channel are secured in the bacterial membranes through a series of highly symmetrical rings [4]. The sorting of the secretion substrates is highly regulated, in space and time in response to the environmental signals received, through a large cytoplasmic platform that gates the secretion core [5][6]. The early secretion substrates build the extracellular parts of these machineries, such as the so-called hollow inner rod that extends to the hollow needle or pilus structure (
). Middle secretion stage substrates then follow. These are, for example, either the helper proteins to break down the plant cell wall and clear the path for the growing pilus, οr the translocator proteins charged with the task to form pores in the host cell membrane. The T3S nanomachinery is then docked to these translocator pores and the secretion of the late secretion substrates, the bacterial Type III Secretion effectors (T3SEs), is finally allowed [7].
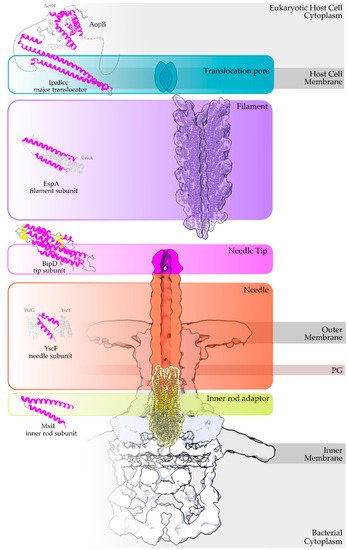
Overview of a T3S nanomachine embedded in the two bacterial membranes. Early and middle secretion substrates are almost all α-helical. The early secretion substrates build the inner rod adaptor (yellow α-helices just above the secretion core, PDB id 6RWY) and the needle (in orange, EMDB id 2803, red α-helices, PDB id 6RWY) or pilus of the nanomachinery that extends to the extracellular space. In animal pathogenic bacteria, the needle is capped with the pentameric needle tip (here in blue, EMDB id 2803), a middle stage substrate. Enteropathogenic (EPEC)/enterohemorrhagic (EHEC)
produce thicker extracellular appendages termed filaments (here in purple, PDB id 7KHW). The rest of the parts of the T3S nanomachine, that are not formed from secretion substrates, are depicted in shades of grey to white (PDB id: 6Q15, EMDB ids: 20561, 20611). In the left column, high resolution structures in cartoon representation (α-helices in magenta, β-strands in yellow) corresponding to early secretion substrates that polymerize to build the corresponding T3S parts shown in the right column. Chaperones to maintain early secretion substrates inside the bacterial cytoplasm have also been described (here represented in different shades of grey: light grey for chaperones with tetratrico-peptide repeats (TPR), grey for the rest). Translocators are also considered middle stage secretion substrates as they must be secreted before the effectors. The major subunit of the translocation pore is maintained inside the bacterial cytoplasm in a secretion competent folding state. The chaperone of the translocator, which also possesses TPR, anchors to the N-terminal Chaperone Binding Domain (CBD) of the translocator, while also covering the transmembrane helices of the translocator by extensively interacting with it [8]. IpaBcc denotes the known long coiled coil domain of IpaB. The CBD is proceeding this domain, while the transmembrane helices are following this domain and shown here protected by the chaperone in an analogy to the AcrH/AopB case [9]. PDB ids used for the left column: 3WXX, 5WKQ, 6RWY, 2IZP, 2P58, 1XOU.
2. Type III Secretion Effectors
Type III Secretion Effectors (T3SEs) are an extremely diverse group of proteins. Bacteria usually possess a huge weaponry targeting a vast array of eukaryotic pathways, with some of them acting even inside the host cell nucleus or other cellular compartments [10]. The main goals are to bypass or trick the eukaryotic defense mechanisms and manipulate the host on behalf of the bacterium [11]. T3SEs mimic many eukaryotic proteins in order to trick the pathways they target [12]. T3SEs do not act individually though—they form robust networks. These networks are flexible enough to tolerate effector losses up to 60% without affecting virulence, while the network composition contributes to host adaptability [13]. A large subgroup of T3SEs targeting animal cells have evolved domains able to reorganize the host cell cytoskeleton in order to (a) promote bacterial uptake, (b) hamper the fusion of the bacterial containing vacuole to lysosomes, or (c) use actin tails to freely move the bacterium inside the host cell cytoplasm, to name a few. On the contrary, plant pathogenic bacteria are obliged to a faster compositional turnover of their T3SE networks to accommodate in addition the R protein-mediated secondary defense [14]. As a result, plant pathogenic bacteria end up with a remarkably higher number of T3SE genes in their genome in comparison to their animal pathogenic counterparts.
3. T3SEs are translocated as unfolded polypeptides
Despite the different evolutionary pressures applied to animal and plant pathogenic bacteria, some common rules are followed. The bacterium has to produce proteins which successfully mimic eukaryotic ones in order to manipulate the host. These new protein domains should be maintained in a secretion competent folding state when inside the bacterial cytoplasm, delivered successfully to the secretion machinery, easily unfolded to pass through the narrow secretion gate, reach the other end and successfully fold after their release to the eukaryotic cell cytoplasm. To fulfill some of these prerequisites, T3SEs possess N-terminal, usually unstructured, regions with specific characteristic biases and patterns [15]. Moreover, when inside the bacterial cytoplasm, the T3SEs usually form complexes with specialized chaperones through their N-terminal, Chaperone Binding Domain (CBD), until the time comes for the delivery of the substrate to the secretion gating mechanism. The T3SS ATPase is used as the energy source for the release of the chaperone and the subsequent unfolding of the T3SE [16]. Interestingly, not all protein domains can be unfolded successfully by the T3SS ATPase. Chimeras of various domains with N-terminal secretion domains of T3SEs are able to block the secretion and trap these chimeric substrates to the secretion channel [17]. The Proton Motive Force (PMF) is considered to be the driving force of the unfolded polypeptide inside the secretion tunnel [18]. In the case of the bacterial flagellum, PMF provides the energy for the flagellar rotation [19]. However, in the T3SS case, PMF has been proposed to facilitate secretion through needle rotation opposite to the right-handed helical grooved surface of the secretion channel [20][21] (
), while alternative theories have also been proposed, such as the contribution from the refolding of the effectors, upon exiting the channel, which could pull forward the following polypeptides [18].
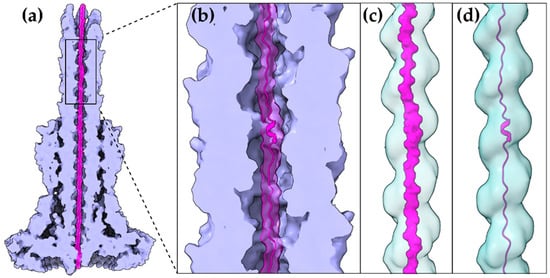
A T3S effector trapped inside the secretion tunnel. The T3SS needle complex atomic structure of
was determined in two functional states. Here with the secretion substrate trapped inside (PDB id 7ahi) [21]. (
) A calculated 5 Å map of the atomic model is shown for simplicity. Substrate is displayed in magenta, T3SS needle complex in light purple. (
) Zoom on the needle and the secretion tunnel. (
,
) Smoothed surface of the tunnel displayed in light blue. The tunnel has a right-handed helical grooved surface and is wide enough to allow secretion substrates to form α-helices. In (
) the molecular surface representation of the substrate is shown. In (
) the cartoon representation is shown.
The gating mechanism for unfolded protein translocation has been revealed recently, as a substrate-engaged needle complex structure, which it has been solved and analyzed in high enough resolution [21]. The far longer secretion tunnel, however, is shaped to a right-handed helix with a minimal inner diameter around 13 Å, a space large enough to accommodate α-helices (
c,d). The substrate density itself, as seen inside the lumen, is comparable to low resolution tubular densities of α-helices, further supporting this view. Although a fully unfolded polypeptide is probably needed to bypass the unidirectional portals found on the secretion core and entrance gate of the T3S machinery, it is possible that α-helices might refold when the polypeptide reaches the needle tunnel (
). The preference towards α-helices might ensure the fast refolding of the substrates when on the other side of the tunnel. This could also be advantageous for a fast-growing needle.
Due to the highest sequence conservation of the needle protomers on their C-terminal part, the part that is directed to the interior of the needle, we can safely hypothesize the existence of a common translocation mechanism adopted by several T3SS families. The
T3S filament is another unique structure, arising from the evolutionary need of the bacterium to penetrate the intestinal mucus layer, which we can gain valuable insights from. This filament also seems to possess a large enough lumen (22 Å) probably capable of forming a seamless conduit with the T3S needle tunnel as both the architecture of the filament lumen and its electrostatic properties resemble the ones of the T3SS needle tunnel [22]. Unfortunately, we lack high resolution data from the much longer pilus (several μm) of the plant pathogenic T3SSs that penetrates the thick plant cell wall. However, it is quite possible that plant associated T3SSs are also following the same, still not fully understood, T3S translocation mechanism.
4. A higher α-helical content is observed in T3SEs.
The α-helix, the most frequently occurring secondary structural element of proteins [23], is a prevalent feature of type III secretion substrates [24][25]. Remarkably, early and middle stage secretion substrates are found to be almost exclusively α-helical (
). In contrast to β-sheets, α-helices are stabilized through hydrogen bonds formed between adjacent, in the sequence, residues. Hence, the preference for α-helices may reflect the need for local hydrogen bonding when the unfolded polypeptide chain reaches the secretion tunnel, where intact α-helices are probably allowed to be formed (
). This permits the secretion substrates to become folding competent as soon as they reach the other end of the secretion tunnel. However, when it comes to T3SEs, the use of β-strands is also observed (
). Despite the occurrence of β-strands, the average preference of T3SEs for α-helices is 10% higher than the average preference in the PDB proteome, while the occurrence of β-strands is approximately 4% lower (
).
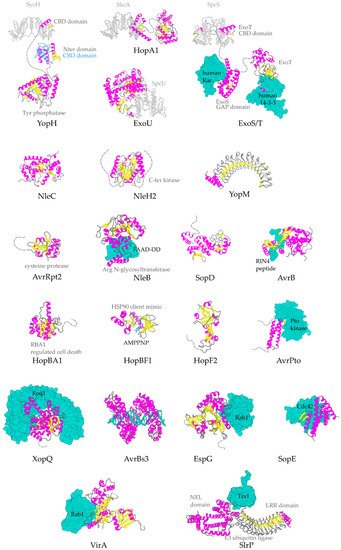
Representative structures for T3SEs. T3SEs are shown in cartoon representation with α-helices in magenta, β-strands in yellow, rest of secondary structure in grey. N-terminal and C-terminal unstructured parts are depicted here in grey broken lines. Dimeric T3SS class I chaperones (or chaperones of effectors) are displayed in grey cartoon representation. Host protein targets that have been co-crystallized with the T3SEs are shown in surface representation in green color. In blue, the alternative conformation of YopH Chaperone Binding Domain (CBD), in the absence of the cognate chaperone. PDB ids used: 1JL5, 4PUF, 4O96, 4O2I, 2QKW, 2NUD, 4FMB, 1GZS, 5CPC, 1S21, 3TU3, 6ACI, 1HE1, 1XXP, 6GNN, 7JLU, 2YPF, 6HQZ, 4RSW, 5T09, 6PWD.
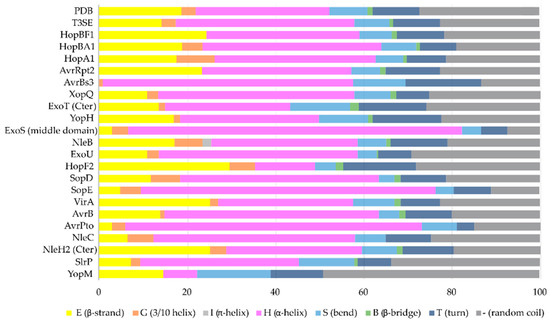
The secondary structure composition of T3SE domains in comparison to PDB. The average occurrence of α-helices in T3SEs is approximately 10% higher compared to the PDB proteome [26]. However, the information from the T3SE-determined crystal structures usually comes from the structured domains of the T3SEs, while their frequently unstructured/flexible N-terminal and C-terminal parts are usually missing from the determined crystal structures. Secondary structure assignments were performed for each PDB id presented in
.
4.1 Gatekeepers: the unique all α-helical, T3SEs
All T3SS gatekeepers are α-helical proteins and, among the T3SEs, the first ones to be translocated. They are conserved proteins encoded usually in the T3S gene cluster, charged with an additional important dual role: they mimic T3SEs by occupying a specific position to the export gate and blocking their secretion, while prioritizing the secretion of translocators at the same time [6][26][27]. Depending on the T3S system they serve, gatekeepers consist of one or two polypeptides [27]. They can be classified as later substrates themselves, not only because they are translocated inside the host cell cytoplasm but also because they use a class I T3S chaperone (chaperone of the effectors) for their delivery to the secretion machinery. However, there is a striking difference: this chaperone is not a homodimer but a heterodimer, probably adding to the complexity of this tightly regulated system [6][27]. Gatekeepers have also been found to bind translocator-specific TPR chaperones with their carboxy terminal part, probably facilitating, in this way, the recruitment of the translocators to the secretion gate [28][29].
The
CopN gatekeeper has been found to inhibit microtubule nucleation when inside the host [30][31]. The whole molecule of CopN is characterized by high plasticity, with both terminal ends completely disordered and absent from the electron density maps of the structures determined by X-ray crystallography. The rest of the protein folds in an elongated tandem repeat of three quite similar five-helix motifs named R1 to R3 (
) [32]. Despite their structural homology, the motifs share low sequence similarity. Each motif is composed of two sets of parallel helices which are packed together in a crossing angle of approximately 50 degrees. The one set comprises a helix–loop–helix pair (yellow in
b) and the other a triplex of helices with one (in motifs R1, R3) or two (in motif R2) of them to be shared between successive motifs. The crystal structures of CopN in complex with Scc3 and tubulin display the fundamental role of R2 and R3 motifs in these interactions (
c,d) [28][30]. The CopN structure resembles the structures of other gatekeepers, such as that of the single chain
MxiC and the heterodimeric
YopN/TyeA.
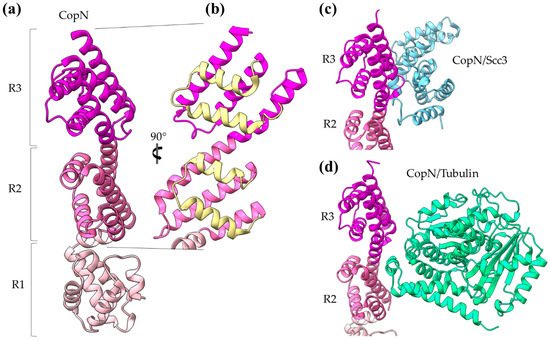
CopN gatekeeper structure. (
) The CopN structure consisting of three (R1–R3) structurally homologous 5-helix assemblies, shown in different magenta shades. (
) Close-up of the R2 and R3 motifs of CopN. The view of panel (
) is rotated 90 degrees in comparison with the view in panel (
). The pair of parallel helices is shown in yellow for clarity. (
) and (
) Part of the CopN structure (only the R3 and R2 motifs are shown) in complex with the Scc3 chaperone of translocators (cyan) and the tubulin (green), respectively.
5. The Diverse Roles of α-Helical Domains
The main advantage of α-helices when compared to β-sheets is the local character of required hydrogen bonding, which stabilizes this secondary structural element. T3SEs are probably allowed to form α-helices in the secretion tunnel after passing the secretion gating mechanism [21]. These pre-folded parts may render the T3SEs highly folding competent by acting as nuclei that promote the rapid overall folding of the polypeptide chain once inside the host cell cytoplasm. α-helical domains may achieve high folding/unfolding rates, as it has been demonstrated in the literature [33][34][35][36][37].
There are multiple ways that the T3SE helical domains support the pathogenic requirements for specific catalytic activity, tight regulation and structural plasticity: (i) Helical assemblies form structural scaffolds from which other elements, for instance, bulges and loops, are protruding and achieve either protein–protein interactions or provide catalytically important residues. (ii) Individual α-helices can be actively involved in the function by, for example, providing catalytic or metal binding residues. (iii) α-helices assemble together to form binding sites of the required physicochemical properties concerning, for example, hydrophobicity or electrostatics.
The 4-α-helix bundles appeared several times in the T3SEs we discussed earlier, serving a variety of functions. We can distinguish two main interaction patterns that this motif uses for function: it either provides an extra element which folds out of the main helical assembly or one of the four main helices is directly involved in the function. The AvrPtoB and the YopE family of T3SE GAPs use bulges and loops to make interactions with their protein partners and even to provide significant residues to interaction sites, whereas the ExoU family uses its 4-α-helix bundle in a dual way (Figure 6). At least one of the loops of the bundle anchors the protein to the membrane, while one of the bundle helices makes partial interactions with a regulatory ubiquitin molecule and contributes to the protein’s activation.
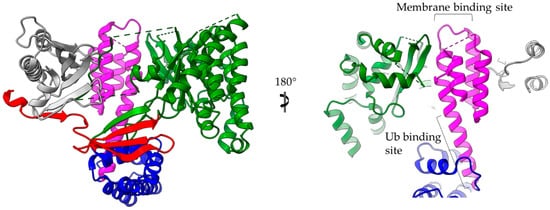
ExoU structure (multi-colored) in complex with the SpcU chaperone (gray). The complex is shown in two views 180 degrees apart. At the right panel the molecule has been clipped for the emphasis to be given on the 4-helix-bundle (magenta). ExoU comprises a highly unstructured N-terminal domain (red), which interacts with the chaperone, a catalytic, patatin-like domain (green), an all-helical bridging domain (blue) and a C-terminal 4-helix domain (magenta). The 4-α-helix-bundle domain is involved in the membrane and ubiquitin binding. The corresponding binding sites are indicated. Ub stands for ubiquitin.
The same concept also applies for other helical assemblies. Indeed, 3- and 2-helix bundles, namely helix–loop–helix motifs and coiled coils, are frequently used by T3SEs. AvrPto uses bulges protruding from its 3-helix bundle to make interactions with the Pto, and the modular TAL effectors use the loops of a repeated helix–loop–helix motif to achieve specific DNA binding (Figure 7). The SopE family of GEFs folds in a novel V-shaped assembly composed of two 3-helix bundles and provides the catalytic residue from one of the three connecting loops. Similarly, the catalytic Cys of the NEL T3SE E3 ligases, which are composed of 12 to 14 helices, resides on a loop connecting two helices.
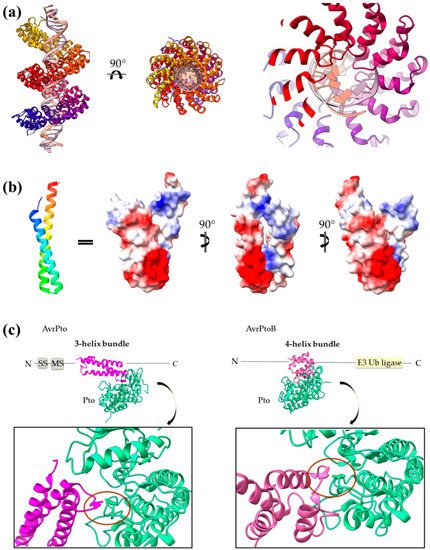
(
) PthXo1 protein in complex with DNA (PDB id 3UGM). The structure comprises tandem repeats of a helix–loop–helix motif. The protein is colored from N-terminus (blue) to C-terminus (red). DNA specific recognition and binding occurs through the hypervariable, in sequence, loop of the motif (right). (
) The AvrRps4 mature structure is a coiled coil with electrostatically diverse sides (PDB id 4B6X). Red and blue colors on the surface denote negative and positive charge, respectively. (
) Each of the AvrPto and AvrPtoB proteins (magenta) interact with the Pto host kinase (green). Left: schematic diagram of the AvrPto protein and close-up of the AvrPto–Pto interaction (PDB id 2QKW). Right: schematic diagram of the AvrPtoB protein and close-up of the AvrPtoB–Pto interaction (PDB id 3HGK).
NEL domains can also provide an example of regulation by α-helices. For at least a member of the family, it has been shown that the loop where the catalytic residue resides can be re-folded in a helix dependent on the redox conditions. More trivial examples of regulation based on helices include T3SEs which possess flexible helical caps able to oscillate around an active/binding site. Helices can also constitute barriers which block and isolate functional sites.
In a different mode of action, the coiled coil of AvrRps4 seems to use the electrostatic properties of its surface to make interactions with protein factors. Likewise, the T3SE zinc metalloproteases mimic DNA electrostatics to create a suitable active site cleft in order to specifically bind their substrate. In addition, these enzymes use an active site helix which provides several residues important for the catalysis. The same is also true for the VirA family of T3SE GAPs we described. The universally used for GAP action Arg residue is provided by an α-helix, which also participates in the substrate binding.In a different mode of action, the coiled coil of AvrRps4 seems to use the electrostatic properties of its surface to make interactions with protein factors. Likewise, the T3SE zinc metalloproteases mimic DNA electrostatics to create a suitable active site cleft in order to specifically bind their substrate. In addition, these enzymes use an active site helix which provides several residues important for the catalysis. The same is also true for the VirA family of T3SE GAPs we described. The universally used for GAP action Arg residue is provided by an α-helix, which also participates in the substrate binding.
References
- Portaliou, A.G.; Tsolis, K.C.; Loos, M.S.; Zorzini, V.; Economou, A. Type III Secretion: Building and Operating a Remarkable Nanomachine. Trends Biochem. Sci. 2016, 41, 175–189.
- Tampakaki, A.P.; Skandalis, N.; Gazi, A.D.; Bastaki, M.N.; Sarris, P.F.; Charova, S.N.; Kokkinidis, M.; Panopoulos, N.J. Playing the “Harp”: Evolution of our understanding of hrp/hrc genes. Annu. Rev. Phytopathol. 2010, 48, 347–370.
- Abby, S.S.; Rocha, E.P.C. The Non-Flagellar Type III Secretion System Evolved from the Bacterial Flagellum and Diversified into Host-Cell Adapted Systems. PLoS Genet. 2012, 8, e1002983.
- Lunelli, M.; Kamprad, A.; Bürger, J.; Mielke, T.; Spahn, C.M.T.; Kolbe, M. Cryo-EM structure of the Shigella type III needle complex. PLoS Pathog. 2020, 16, e1008263.
- Hu, B.; Morado, D.R.; Margolin, W.; Rohde, J.R.; Arizmendi, O.; Picking, W.L.; Picking, W.D.; Liu, J. Visualization of the type III secretion sorting platform of Shigella flexneri. Proc. Natl. Acad. Sci. USA 2015, 112, 1047–1052.
- Charova, S.N.; Gazi, A.D.; Mylonas, E.; Pozidis, C.; Sabarit, B.; Anagnostou, D.; Psatha, K.; Aivaliotis, M.; Beuzon, C.R.; Panopoulos, N.J.; et al. Migration of type III secretion system transcriptional regulators links gene expression to secretion. mBio 2018, 9.
- Deng, W.; Marshall, N.C.; Rowland, J.L.; McCoy, J.M.; Worrall, L.J.; Santos, A.S.; Strynadka, N.C.J.; Finlay, B.B. Assembly, structure, function and regulation of type III secretion systems. Nat. Rev. Microbiol. 2017, 15, 323–337.
- Ferrari, M.L.; Charova, S.N.; Sansonetti, P.J.; Mylonas, E.; Gazi, A.D. Structural Insights of Shigella Translocator IpaB and Its Chaperone IpgC in Solution. Front. Cell. Infect. Microbiol. 2021, 11, 353.
- Nguyen, V.S.; Jobichen, C.; Tan, K.W.; Tan, Y.W.; Chan, S.L.; Ramesh, K.; Yuan, Y.; Hong, Y.; Seetharaman, J.; Leung, K.Y.; et al. Structure of AcrH-AopB Chaperone-Translocator Complex Reveals a Role for Membrane Hairpins in Type III Secretion System Translocon Assembly. Structure 2015, 23, 2022–2031.
- Li, G.; Froehlich, J.E.; Elowsky, C.; Msanne, J.; Ostosh, A.C.; Zhang, C.; Awada, T.; Alfano, J.R. Distinct Pseudomonas type-III effectors use a cleavable transit peptide to target chloroplasts. Plant J. 2014, 77, 310–321.
- Parsot, C. Shigella type III secretion effectors: How, where, when, for what purposes? Curr. Opin. Microbiol. 2009, 12, 110–116.
- De Groot, N.S.; Burgas, M.T. Bacteria use structural imperfect mimicry to hijack the host interactome. bioRxiv 2020, 16, e1008395.
- Ruano-Gallego, D.; Sanchez-Garrido, J.; Kozik, Z.; Núñez-Berrueco, E.; Cepeda-Molero, M.; Mullineaux-Sanders, C.; Clark, J.N.B.; Slater, S.L.; Wagner, N.; Glegola-Madejska, I.; et al. Type III secretion system effectors form robust and flexible intracellular virulence networks. Science 2021, 371.
- McCann, H.C.; Guttman, D.S. Evolution of the type III secretion system and its effectors in plant-microbe interactions. New Phytol. 2007, 177, 33–47.
- Cunnac, S.; Lindeberg, M.; Collmer, A. Pseudomonas syringae type III secretion system effectors: Repertoires in search of functions. Curr. Opin. Microbiol. 2009, 12, 53–60.
- Akeda, Y.; Galán, J.E. Chaperone release and unfolding of substrates in type III secretion. Nature 2005, 437, 911–915.
- Radics, J.; Königsmaier, L.; Marlovits, T.C. Structure of a pathogenic type 3 secretion system in action. Nat. Struct. Mol. Biol. 2014, 21, 82–87.
- Lee, P.C.; Rietsch, A. Fueling type III secretion. Trends Microbiol. 2015, 23, 296–300.
- Nakamura, S.; Minamino, T. Flagella-Driven Motility of Bacteria. Biomolecules 2019, 9, 279.
- Ohgita, T.; Saito, H. Biophysical mechanism of protein export by bacterial type III secretion system. Chem. Pharm. Bull. 2019, 67, 341–344.
- Miletic, S.; Fahrenkamp, D.; Goessweiner-Mohr, N.; Wald, J.; Pantel, M.; Vesper, O.; Kotov, V.; Marlovits, T.C. Substrate-engaged type III secretion system structures reveal gating mechanism for unfolded protein translocation. Nat. Commun. 2021, 12, 1546.
- Zheng, W.; Peña, A.; Ilangovan, A.; Clark, J.N.B.; Frankel, G.; Egelman, E.H.; Costa, T.R.D. Cryoelectron-microscopy structure of the enteropathogenic Escherichia coli type III secretion system EspA filament. Proc. Natl. Acad. Sci. USA 2021, 118.
- Eisenberg, D. The discovery of the α-helix and β-sheet, the principal structural features of proteins. Proc. Natl. Acad. Sci. USA 2003, 100, 11207–11210.
- Gazi, A.D.; Charova, S.N.; Panopoulos, N.J.; Kokkinidis, M. Coiled-coils in type III secretion systems: Structural flexibility, disorder and biological implications. Cell. Microbiol. 2009, 11, 719–729.
- Charova, S.N.; Gazi, A.D.; Kotzabasaki, M.; Sarris, P.F.; Fadouloglou, V.E.; Panopoulos, N.J.; Kokkinidis, M. Protein Flexibility and Coiled-Coil Propensity: New Insights Into Type III and Other Bacterial Secretion Systems. In Biochemistry; InTech: London, UK, 2012.
- Micsonai, A.; Wien, F.; Kernya, L.; Lee, Y.-H.; Goto, Y.; Réfrégiers, M.; Kardos, J. Accurate secondary structure prediction and fold recognition for circular dichroism spectroscopy. Proc. Natl. Acad. Sci. USA 2015, 112, E3095–E3103.
- Schubot, F.D.; Jackson, M.W.; Penrose, K.J.; Cherry, S.; Tropea, J.E.; Plano, G.V.; Waugh, D.S. Three-dimensional structure of a macromolecular assembly that regulates type III secretion in Yersinia pestis. J. Mol. Biol. 2005, 346, 1147–1161.
- Archuleta, T.L.; Spiller, B.W. A Gatekeeper Chaperone Complex Directs Translocator Secretion during Type Three Secretion. Plos Pathog. 2014, 10, e1004498.
- Cherradi, Y.; Schiavolin, L.; Moussa, S.; Meghraoui, A.; Meksem, A.; Biskri, L.; Azarkan, M.; Allaoui, A.; Botteaux, A. Interplay between predicted inner-rod and gatekeeper in controlling substrate specificity of the type III secretion system. Mol. Microbiol. 2013, 87, 1183–1199.
- Campanacci, V.; Urvoas, A.; Cantos-Fernandes, S.; Aumont-Nicaise, M.; Arteni, A.A.; Velours, C.; Valerio-Lepiniec, M.; Dreier, B.; Plückthun, A.; Pilon, A.; et al. Insight into microtubule nucleation from tubulin-capping proteins. Proc. Natl. Acad. Sci. USA 2019, 116, 9859–9864.
- Huang, J.; Lesser, C.F.; Lory, S. The essential role of the CopN protein in Chlamydia pneumoniae intracellular growth. Nature 2008, 456, 112–115.
- Nawrotek, A.; Guimarães, B.G.; Velours, C.; Subtil, A.; Knossow, M.; Gigant, B. Biochemical and structural insights into microtubule perturbation by CopN from Chlamydia pneumoniae. J. Biol. Chem. 2014, 289, 25199–25210.
- Taylor, J.W.; Greenfield, N.J.; Wu, B.; Privalov, P.L. A calorimetric study of the folding-unfolding of an α-helix with covalently closed N and C-terminal loops. J. Mol. Biol. 1999, 291, 965–976.
- Ihalainen, J.A.; Paoli, B.; Muff, S.; Backus, E.H.G.; Bredenbeck, J.; Woolley, G.A.; Caflisch, A.; Hamm, P. α-Helix folding in the presence of structural constraints. Proc. Natl. Acad. Sci. USA 2008, 105, 9588–9593.
- Berkemeier, F.; Bertz, M.; Xiao, S.; Pinotsis, N.; Wilmanns, M.; Gräter, F.; Rief, M. Fast-folding α-helices as reversible strain absorbers in the muscle protein myomesin. Proc. Natl. Acad. Sci. USA 2011, 108, 14139–14144.
- Schennach, M.; Schneeberger, E.M.; Breuker, K. Unfolding and Folding of the Three-Helix Bundle Protein KIX in the Absence of Solvent. J. Am. Soc. Mass Spectrom. 2016, 27, 1079–1088.
- Jesus, C.S.H.; Cruz, P.F.; Arnaut, L.G.; Brito, R.M.M.; Serpa, C. One Peptide Reveals the Two Faces of α-Helix Unfolding-Folding Dynamics. J. Phys. Chem. B 2018, 122, 3790–3800.