Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Petros Christopoulos and Version 2 by Rita Xu.
The principles and current clinical landscape of multispecific antibodies against cancer.
- bispecific antibodies
- multispecific antibodies
- monoclonal antibodies
- therapeutic antibodies
1. Introduction
The tortuous, 2.5 billion-years-long path from immunoglobulin (Ig)-like domains of archaeal flagellins to the antibodies (Ab) of jawed vertebrates is one of the most intriguing discoveries in evolutionary biology [1][2][3][1,2,3]. Equally impressive are the accomplishments of modern genetic engineering, whose further variation of basic Ig building blocks could produce over 40 different molecular formats of therapeutic antibodies during the last two decades [4]. As times change, priorities shift, and pathogen defense has today largely been succeeded by a much more challenging task: the fight against cancer [5].
The concept that antibodies could be used as “magic bullets” against human maladies dates back to their discovery in the late 19th century [6] and the gradual recognition that they can bind a virtually unlimited number of antigens with a high specificity and affinity [7]. However, it was not until discovery of the “hybridoma” technology in 1975 [8], complemented by various humanization techniques a few years later [9], that scientists managed to harness this power: monoclonal antibodies could now be produced in large quantities after injecting a mouse (later, rat or other mammal) with the desired antigen, and fusing the respective splenocytes with suitable myeloma cell lines [10]. In the next step, two hybridomas were fused (“hybrid hybridoma”, aka “quadroma”) to produce bispecific antibodies without the protein denaturation steps necessary for chemical cross-linking, which could potentially adversely affect binding properties [11][12][11,12].
Improved function is the main incentive behind development of multispecific constructs. For every antibody, the “classic” mode of action falls into two broad categories: (i) “disruptive” with respect to the epitope-bearing target molecule, i.e., blocking or activating signals, neutralizing antigens, or causing internalization and degradation of surface receptors and (ii) “recruiting”, i.e., activating immune cells and/or other effector molecules, like the complement [13]. OKT3 (aka “muromonab-CD3”), for example, the first monoclonal antibody to ever achieve regulatory approval in 1986, is a typical product of the first category, used to suppress T-cell function in patients with glucocorticoid-resistant rejection of allogeneic renal, heart and liver transplants [14]. Rituximab, on the other hand, a CD20-specific monoclonal antibody still widely used since its approval in 1997, kills B- cells by combining signaling-induced death with cellular and complement-mediated cytotoxicity [15]. Compared to monospecific monoclonal antibodies, multispecific constructs potentiate antibody-mediated effects, for example, they can potentially “disrupt” multiple instead of one tumor-associated antigens (TAA) owing to more antigen-binding regions, and/or they can “recruit” and activate immune cells even stronger, since they use dedicated antigen-binding sites for this. The functional augmentation facilitated by multispecificity is clinically relevant: it translates into improved response rates, for example, approximately 50% with the newer anti-CD20/CD3 bispecific antibodies as monotherapy in B-cell non-Hodgkin’s lymphomas (B-NHL) which do not respond to rituximab any more [16], can delay development of resistance, and simplifies drug development compared to the more complicated, expensive and time-consuming procedures necessary for launching of multiple monospecific products instead [4].
Wide adoption of genetic engineering facilitates today’s exploitation of the huge potential inherent in multispecific antibodies: suitable polypeptide chains are designed in silico and expressed in various host systems, most frequently CHO cells and E. coli, followed by purification and assembly of the various components in vitro [17][18][19][17,18,19]. Appropriate antigen-binding properties, high yield, high thermal and chemical stability, good solubility, and low viscosity have key importance for large-scale production and clinical applicability [20][21][20,21]. The biochemical basis of these characteristics and our ability to manipulate them lie rooted in the modular antibody structure (Figure 1a).
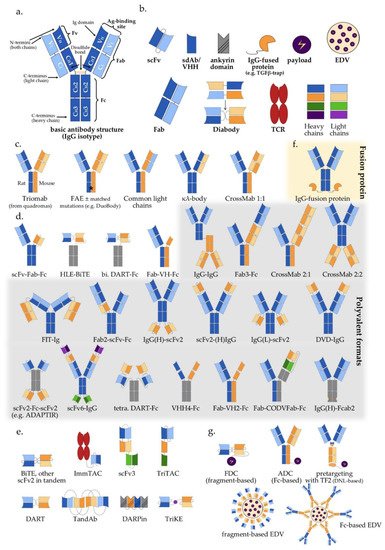
Figure 1. Multispecific antibody formats in clinical trials: (a) Basic IgG structure; (b) Main antibody components (detailed in Section 2 of the main text); (c) IgG-like “two-halves” bispecific formats (following the order of Section 3.1 and Table 1); (d) IgG-modified (appended and/or substituted, Fc-based) bispecific and multispecific antibodies (Table 1); (e) Fragment-based (Fc-free) bispecific and multispecific antibodies (Table 1); (f) IgG fusion protein; (g) Bi-/multispecific constructs for payload delivery (Table 1). For the abbreviations, please see the respective section in the main text.
Table 1. Multispecific antibody formats in clinical trials by structure, mode of action and start year of first study.
| Class | Specificity/ Valence |
Specificity/ Valence |
Action (C/R) | Format | No. of Clinical Trials | Comment | First Clinical Trial |
|---|
| Fc-based | IgG-like (Figure 1c) | (Section 3.1) (Figure 1c) |
2/2 | R | TrioMab | 9 | “two-halves” formats |
2004 | ||
| 2/2 | C/R | FAE | 36 | 2010 | ||||||
| 2/2 | C/R | common light chain | 19 | 2014 | ||||||
| 2/2 | C | κλ-body | 2 | 2019 | ||||||
| 2/2 | C/R | CrossMab 1:1 | 12 | 2012 | ||||||
| IgG-modified (Figure 1d) | (Section 3.2 & Section 4) (Figure 1d) |
2/2 | C/R | scFv-Fab-Fc | 18 | scFv-monosubstituted | 2016 | |||
| 2/2 | R | HLE-BiTE | 11 | scFv-bisubstituted | 2015 | |||||
| 2/2 | R | DART-Fc | 2 | Db-bisubstituted | 2014 | |||||
| 2/2 | R | Fab-VH-Fc ** | 3 | V | H | -monosubstituted | 2021 | |||
| 2/4 | R | IgG-IgG | 15 | IgG-IgG | 2004 | |||||
| 2/3 | R | Fab3-Fc | 1 | Fab-appended | 2018 | |||||
| 2/3 | R | CrossMab 2:1 | 10 | 2014 | ||||||
| 2/4 | C | CrossMab 2:2 | 1 | 2015 | ||||||
| 2/4 | C/R | FIT-Ig (Fabs-in-Tandem) | 3 | 2018 | ||||||
| 2/3 | R | Fab2-scFv-Fc | 2 | scFv-appended | 2020 | |||||
| 2/4 | C/R | IgG(H)-scFv2 | 19 | 2017 | ||||||
| 2/4 | C | scFv2-(H)IgG | 1 | 2014 | ||||||
| 2/4 | R | IgG(L)-scFv2 | 2 | 2019 | ||||||
| 2/4 | C | DVD-IgG | 1 | V-appended | 2013 | |||||
| 2/4 | R | scFv2-Fc-scFv2 | 2 | scFv-multisubstituted | 2015 | |||||
| 4/8 | R | scFv6-IgG | 2 | 2020 | ||||||
| 2/4 | C | DART-Fc | 3 | Db-multisubstituted | 2017 | |||||
| 2/4 | C | VHH4-Fc | 5 | V-multi substituted | 2019 | |||||
| 2/3 | C/R | Fab-VH2-Fc ** | 4 | 2019 | ||||||
| 3/3 | R | Fab-CODVFab-Fc | 1 | 2020 | ||||||
| 2/3-4 * | C | IgG-fusion proteins | 55 | Fusion moiety | 2015 | |||||
| 2/4 | C | IgG(H)-Fcab2 | 3 | Fc-modified | 2018 | |||||
| Fc-free | Fv-based $ | ( | $ Figure 1e) | Section 3.3 & Section 4) ($ Figure 1e) |
2/2 | R | BiTE | 20 | scFv-based | 2008 |
| 2/2 | C/R | other scFv2 in tandem | 6 | 2005 | ||||||
| 2/2 * | R | ImmTAC | 5 | 2015 | ||||||
| 3/3 * | R | TriKE | 1 | 2020 | ||||||
| 3/3 | C | scFv3 | 1 | 2020 | ||||||
| 2/2 | R | DART | 6 | Db-based | 2014 | |||||
| 2/4 | R | TandAb | 6 | 2010 | ||||||
| 3/3 | R | TriTAC | 4 | 2018 | ||||||
| ¾ * | C | DARPin | 5 | Ankyrin-based | 2014 | |||||
| Fab-based | $ | 2/2 | C | Fab2 | 2 | Fab-based | 1997 | |||
| with payload (Figure 1f) | (Section 5) (Figure 1f) |
2/2 # | C | Fc-based ADC/EDV | 4 | IgG-based | 2014 | |||
| 2/2 # | C | Fc-free FDC/EDV | 3 | Fragment-based | 2013 | |||||
| 2/2-3 (#) | C | ±pretargeting (±imaging) | 4 | 2004 | ||||||
Formats are ordered as in FigureFigure 1 1 and the corresponding sections of the main text; C: classical mode of action; R: immune-cell redirecting; for the explanation of other abbreviations, please see the main text; * one binding site does not rely on typical antigen-antibody interaction; ** human VH or VHH; # with payload; $ Figure 1e.
2. Antibody Structure and Approaches to Multispecificity
The typical structure of human antibodies is represented by the IgG isotype, which is the most prevalent class [22]. X-ray crystallographic and electron-microscopic studies have revealed this to be a heterotetrametric, roughly Y-shaped protein with axial symmetry, which consists of two identical heavy (“H”, approximately 50 kDa each), and two identical “light” (“L”, approximately 25 kDa each) polypeptide chains, linked together by disulfide bonds (Figure 1a) [23][24][25][23,24,25]. Basic building block for both chain types is the “Ig domain”, aka “Ig fold”, which is a sandwich-like structure formed by two sheets of 7–9 antiparallel β-strands arranged with a Greek-key topology [26]. This basic motif appears to be conserved throughout the evolution of life, presumably because its efficient and compact folding provides a suitable substrate for numerous essential recognition, binding and adhesion processes carried out by members of the large Ig protein superfamily [1]. Each human antibody heavy chain consists of four domains, three constant ones (termed CH1, CH2 and CH3) and one variable (VH), while the light chains consist of one constant (CL) and one variable domain (VL) each. Limited digestion with the cysteine protease papain splits the IgG antibody in three equal-sized portions, namely two antigen-binding fragments (Fab), each consisting of one light chain bound with the VH and CH1 domains of its partner heavy chain, and one crystallizable fragment (Fc), which contains the remaining constant domains of the heavy chain (CH2 and CH3, Figure 1a) [27]. Within the Fab region, the side-by-side arrangement of the VL and VH domains brings discrete amino acid loops between their β-sheets (“complementarity-determining regions”, CDR) together to form the antigen-binding site at the outer tip (Figure 1a). Direct linking of VL and VH by a peptide chain creates the single-chain variable fragment (scFv), an artificial construct that can also effectively bind antigens (Figure 1b) [28]. Interestingly, the antigen specificity of most antibodies shows a predominant dependence on the CDRs contributed by mainly the VH rather than the VL domain, which takes an extreme form in the heavy chain-only antibodies naturally produced by camelids and sharks [29], and is exploited by “single-domain antibodies” (sdAb), also referred to as nanobodies, consisting of a single VHH (variable heavy chain homodimer) thereof (Figure 1b) [30].
Due to its axial symmetry, the IgG can bind two identical epitopes with two binding sites (“paratopes”), one on each Fab arm, i.e., it is “monospecific”, but “bivalent” [31]. Generally, the valency of an antibody refers to the total number of epitopes that it can bind, and its specificity to the number of different structures among them. Naturally occurring antibodies are generally monospecific, which allows them to cross-link large numbers of antigen molecules and thus amplify the immune response against them [22]. Nonetheless, for IgG4 and possibly also other Ig subclasses, reducing conditions in the blood or cell surfaces can break up disulfide bonds and facilitate Fab-arm exchange (FAE) which gives chimeric bispecific antibodies with anti-inflammatory activity (Figure 1c) [32][33][32,33]. Generation of the first artificial bispecific antibodies in 1961 mimicked this naturally occurring FAE: by reducing and reoxidizing the F(ab’)2 fragments derived by peptic digestion of two different antibodies, their univalent Fab were recombined into new F(ab’)2 molecules that could precipitate a mixture of their cognate antigens, but neither of them in pure form [34][35][34,35]. After 1975, “quadromas” became a relatively easy method to generate full IgG-like bispecific molecules (Figure 1c), but the low yield of the desired antibody (12.5% with random pairing of heavy and light chains), together with the difficulty to isolate it from the closely related mispaired contaminants, remained a significant problem [36]. Over the following decades, many different methods within the constraints of the classical “IgG-like” format were devised to overcome or circumvent the “mispairing” problem of heavy and light chains produced by quadromas or genetic engineering (Figure 1c): rat/mouse quadromas, which took advantage of the species-restricted heavy/light chain pairing and the differential affinity of protein A for mouse and rat heavy chains (“TrioMabs”) [37][38][37,38]; various “knobs-into-holes” (KiH) or other techniques of inducing complementary mutations in the sequences of heavy and/or light chains in order to force the desired heterodimerization [39][40][41][42][39,40,41,42] or facilitate controlled FAE (“Duobodies”) [43]; “common-light-chain” antibodies, in which the two distinct paratopes on each Fab arm utilize the same light chain paired with a different heavy chain in order to bind its target antigen [44]; “κ-λ” bodies, which utilize the same heavy, but different light chains for the two paratopes, in order to obviate the need for artificial mutations or linkers that may result in poor stability and increased potential immunogenicity [45]; “CrossMabs”, with swapping of either the variable or the constant domains between light and heavy chains to create two asymmetric Fab arms that force the desired light chain pairing, while preserving the binding properties of the respective paratope [46][47][46,47]; electrostatic steering effects [48][49][48,49]; IgG/A chimeras, aka “strand exchange engineered domain bodies” (“SEEDBodies”) [50]; the proprietary “Azymetric” heterodimeric Fc [51]; “dual action Fab” (DAF, aka “two-in-one” antibodies), which use the same heavy and light chains to recognize two unrelated antigens via differential use of their two paratopes [52][53][52,53]; “DutaMabs” or “DutaFabs”, in which each Fab arm contains two different paratopes, each utilizing only 3 out of the 6 available CDRs [54].
“IgG-modified” formats (Figure 1d) provide additional solutions to this problem, e.g., in “dual-variable-domain” (DVD) antibodies, each chain contains two variable domains, so that bispecificity is ensured irrespective of light chain pairing [55]. Moreover, using genetic engineering, the antigen-binding moieties Fab, Fv and VHH can be combined freely with each other, or linked to IgGs, resulting in huge structural variability (Figure 1d,e and Table 1) [4][56][4,56]. In addition, monospecific and bispecific formats can be combined in order to increase specificity, valency, or both; for example a highly active tetravalent and tetraspecific “four-in-one” antibody against EGFR, HER2, HER3 and VEGF was generated by combining the DVD, CrossMab and KiH technologies [57]. It should also be noted that the functionality of multispecifics can further be extended through fusion with other non-Ig proteins; for example, the T-cell receptor (TCR) [58], the TGFβ receptor [59], an IL-15 moiety in case of Trispecific Killer Cell Engagers (TriKEs) [60], various payloads [61], and by conjugation with Engeneic Delivery Vehicles (EDV), i.e., bacterially-derived nanocells coated with bispecific antibodies for the targeted delivery of cytotoxics, siRNA and other cargo to the tumor cells (Figure 1f) [62][63][62,63].
Among the numerous possible variations, the single most important structural characteristic of multispecific antibodies is whether they contain an Fc region or not. Fc-based constructs are generally larger, have longer half-lives (typically a few weeks) due to recycling by the neonatal Fc receptor (FcRn) and glomerular preservation [64][65][64,65], show improved solubility and stability, trigger cytotoxic [66] and T-cell priming effects [67], and can be purified using established affinity chromatography workflows [68]. In contrast, Fc-free, antibody fragment-based constructs are usually rapidly cleared from the blood (within minutes), which necessitates either administration by continuous infusion, or fusion with carrier moieties, such as human serum albumin (HSA) or polyethylene glycol (PEG), in order to extend their half-lives [69][70][71][69,70,71]. At the same time, specific advantages of smaller multispecifics can be better diffusion into the tumor tissue, higher potency due to closer proximity of interactions in the two paratopes, ease of large-scale production in microbial systems, and less immune-related adverse effects due to lack of Fc [72][73][74][72,73,74].
In terms of functionality, the basic distinction is between the “classic” and the “recruiting” or “redirecting” mode of antibody action, in which at least one binding site engages invariable immune cell receptors, for example, CD3 on T, or CD16 on NK cells. The combined structural (“Fc-based” vs. “Fc-free”) and functional (“classic” vs. “recruiting”) characterization is a useful framework to contextualize multispecific constructs currently used in clinical trials (Table 1). A detailed description of all constructs and corresponding studies can be found in the full text and supplements of the original IJMS publication.