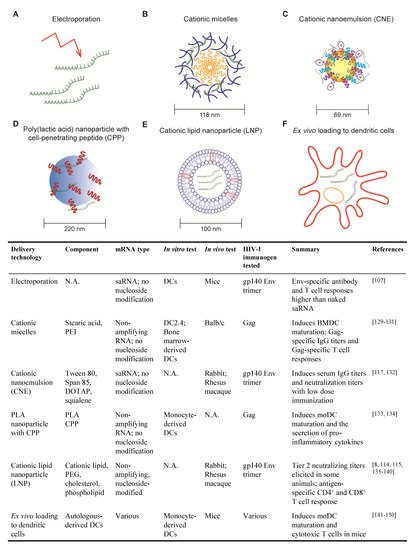
The SARS-CoV-2 pandemic introduced the world to a new type of vaccine based on mRNA encapsulated in lipid nanoparticles (LNPs). Instead of delivering antigenic proteins directly, an mRNA-based vaccine relies on the host’s cells to manufacture protein immunogens which, in turn, are targets for antibody and cytotoxic T cell responses. mRNA-based vaccines have been the subject of research for over three decades as a platform to protect against or treat a variety of cancers, amyloidosis and infectious diseases. In this review, we discuss mRNA-based approaches for the generation of prophylactic and therapeutic vaccines to HIV. We examine the special immunological hurdles for a vaccine to elicit broadly neutralizing antibodies and effective T cell responses to HIV. Lastly, we outline an mRNA-based HIV vaccination strategy based on the immunobiology of broadly neutralizing antibody development.
- HIV
- vaccine
- messenger RNA
1. Progress in mRNA Technology for HIV Vaccines
1.1. Non-Amplifying mRNA Vaccines