Pathogens are one of the main selective pressures that ancestral humans had to adapt to. Components of the immune response system have been preferential targets of natural selection in response to such pathogen-driven pressure. In turn, there is compelling evidence showing that positively selected immune gene variants conferring increased resistance to past or present infectious agents are today associated with increased risk for autoimmune or inflammatory disorders but decreased risk of cancer, the other side of the same coin. CD5 and CD6 are lymphocytic scavenger receptors at the interphase of the innate and adaptive immune responses since they are involved in both: (i) microbial-associated pattern recognition; and (ii) modulation of intracellular signals mediated by the clonotypic antigen-specific receptor present in T and B cells (TCR and BCR, respectively).
- scavenger receptors
- CD5
- CD6
- immune response
- cancer
- autoimmunity
- infections
- natural selection
- human genetics
- single nucleotide polymorphisms
Note: The following contents are extract from your paper. The entry will be online only after author check and submit it.
1. Evolution and Selective Pressure on Immune Receptor Genes: Examples of Selection
1.1. Human Evolution and Pathogens
1.2. Detecting Local Adaptation in the Human Genome
1.3. Examples of Positive Selection at Immune Response Receptors
2. The Case of Lymphocyte Scavenger Receptors CD5 and CD6
2.1. The CD5 and CD6 Protein Receptors: Structure and Function
2.2. The CD5 and CD6 Genes: Location, Exon/Intron Organization and Isoforms
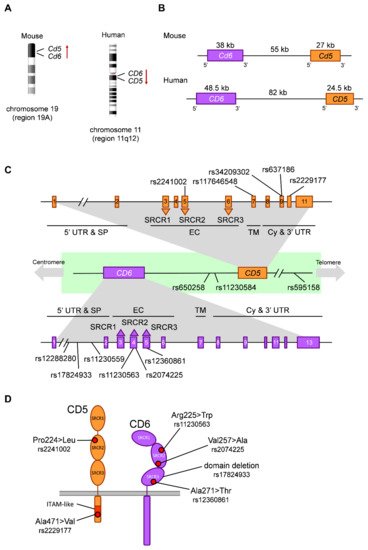
2.3. Functionally Relevant CD5 and CD6 Polymorphisms
2.4. Discovery of the CD5 and CD6 Loci as Targets of Natural Selection
2.5. CD5 Polymorphism in Autoimmunity and Cancer
2.6. CD6 Polymorphism in Autoimmunity and Cancer
| Gene | SNP | Alleles * | Change | CADD | AFR | EUR | EAS | SAS | AMR | Functional/Clinical Relevance | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CD5 | rs2241002 | C>T | Pro224>Leu | 11.06 | 0.31 | 0.15 | 0.06 | 0.15 | 0.14 | T allele associated to lower risk of lupus nephritis | [66] | and higher melanoma mortality | [73] | . Haplotypic combinations with rs2229177 associated to lupus nephritis | [66] | and survival in melanoma | [73] | and chronic lymphocytic leukemia (CLL) | [75] | . | |
| rs117646548 | G>A | Ala377>Thr | 11.86 | 0.00 | 0.01 | 0.00 | 0.00 | 0.01 | |||||||||||||
| rs34209302 | C>T | His461>Tyr | 0.092 | 0.08 | 0.01 | 0.00 | 0.08 | 0.01 | |||||||||||||
| rs637186 | G>A | Arg461>His | 0.014 | 0.01 | 0.08 | 0.00 | 0.04 | 0.05 | |||||||||||||
| rs2229177 | C>T | Ala471>Val | 25.2 | 0.51 | 0.55 | 0.99 | 0.80 | 0.66 | T allele associated to more signaling upon | CD5 | stimulation | [65] | , stronger TCR inhibition | [66] | , decreased lupus nephritis risk | [66] | and lower survival in melanoma | [73] | and CLL | [75] | . |
| Inter-genic | rs650258 | T>C | 0.051 | 0.65 | 0.63 | 0.88 | 0.76 | 0.76 | C allele associated to increased multiple sclerosis (MS) risk | [67][79] | . | ||||||||||
| rs11230584 | G>A | 1.789 | 0.25 | 0.15 | 0.13 | 0.18 | 0.11 | Modulation of | CD5 | and | CD6 | expression | [69] | . | |||||||
| rs595158 | C>A | 2.165 | 0.55 | 0.54 | 0.99 | 0.79 | 0.67 | Risk locus in rheumatoid arthritis | [90] | . | |||||||||||
| CD6 | rs12288280 | G>T | Intronic | 2.973 | 0.50 | 0.10 | 0.10 | 0.05 | 0.14 | T allele associated to decreased neuromyelitis optica risk in an Asian cohort | [84] | . | |||||||||
| rs17824933 | C>G | Intronic | 7.58 | 0.01 | 0.23 | 0.03 | 0.07 | 0.12 | G allele associated to increased expression of | CD6 | Δd3 | [68] | , increased MS risk in European cohorts | [76][77][78] | and increased psoriasis severity | [85] | . | ||||
| rs11230559 | T>C | Intronic | 4.239 | 0.01 | 0.25 | 0.04 | 0.07 | 0.12 | In linkage disequilibrium with rs17824933 | [67] | . | ||||||||||
| rs11230563 | C>T | Arg225>Trp | 22.4 | 0.61 | 0.36 | 0.17 | 0.21 | 0.30 | Haplotypic combinations with rs2074225 associated to differential | CD6 | expression | [67] | . T allele associated to decreased MS risk in an African American cohort | [83] | , decreased psoriasis severity | [85] | and increased Behçet’s disease risk in a Han population | [86] | . Involvement in inflammatory bowel disease | [87][88] | . |
| rs2074225 | T>C | Val257>Ala | 17.66 | 0.33 | 0.38 | 0.59 | 0.54 | 0.56 | Haplotypic combinations with rs11230563 associated to differential | CD6 | expression | [67] | . T allele associated to increased MS risk in a European cohort | [67] | . | ||||||
| rs12360861 | G>A | Ala271>Thr | 0.001 | 0.04 | 0.19 | 0.00 | 0.05 | 0.12 | A allele associated to decreased MS risk in a European cohort | [80] | and increased psoriasis severity | [85] | . |
3. Concluding Remark
Pathogens have exerted strong selective pressures during human evolution, shaping
human immunogenetics. This has resulted in the selection of genetic variants affecting
immune function. CD5 and CD6 are multifaceted lymphocyte scavenger receptors, combining
roles as immune response modulators and pattern recognition receptors. As such,
evolutionarily selected and/or functionally relevant polymorphisms in the CD5 and CD6
loci have been shown to impact a wide variety of immune-related disorders such as autoimmunity
and cancer, often considered two sides of the same coin. This not only reflects
the relevance of genetic variation in the immune function, but also positions CD5 and
CD6 as potentially useful diagnostic and prognostic disease markers, as well as targets of
immunomodulatory therapies.
References
- Cohen, M.N.; Armelagos, G. Paleopathology at the Origins of Agriculture; Cohen, M., Armelagos, G., Eds.; Academic Press: Orlando, FL, USA, 1984; ISBN 978-0-8130-4489-7.
- Morens, D.M.; Fauci, A.S. Emerging Pandemic Diseases: How We Got to COVID-19. Cell 2020, 182, 1077–1092.
- Domínguez-Andrés, J.; Netea, M.G. Impact of Historic Migrations and Evolutionary Processes on Human Immunity. Trends Immunol. 2019, 40, 1105–1119.
- Barreiro, L.B.; Quintana-Murci, L. From evolutionary genetics to human immunology: How selection shapes host defence genes. Nat. Rev. Genet. 2010, 11, 17–30.
- Okada, H.; Kuhn, C.; Feillet, H.; Bach, J.F. The “hygiene hypothesis” for autoimmune and allergic diseases: An update. Clin. Exp. Immunol. 2010, 160, 1–9.
- Akey, J.M. Constructing genomic maps of positive selection in humans: Where do we go from here? Genome Res. 2009, 19, 711–722.
- Sabeti, P.C.; Schaffner, S.F.; Fry, B.; Lohmueller, J.; Varilly, P.; Shamovsky, O.; Palma, A.; Mikkelsen, T.S.; Altshuler, D.; Lander, E.S. Positive natural selection in the human lineage. Science 2006, 312, 1614–1620.
- Williamson, S.H.; Hubisz, M.J.; Clark, A.G.; Payseur, B.A.; Bustamante, C.D.; Nielsen, R. Localizing recent adaptive evolution in the human genome. PLoS Genet. 2007, 3, 0030090.
- Grossman, S.R.; Shlyakhter, I.; Shylakhter, I.; Karlsson, E.K.; Byrne, E.H.; Morales, S.; Frieden, G.; Hostetter, E.; Angelino, E.; Garber, M.; et al. A composite of multiple signals distinguishes causal variants in regions of positive selection. Science 2010, 327, 883–886.
- Fan, S.; Hansen, M.E.B.; Lo, Y.; Tishkoff, S.A. Going global by adapting local: A review of recent human adaptation. Science 2016, 354, 54–59.
- Aguirre-Gamboa, R.; Joosten, I.; Urbano, P.C.M.; van der Molen, R.G.; van Rijssen, E.; van Cranenbroek, B.; Oosting, M.; Smeekens, S.; Jaeger, M.; Zorro, M.; et al. Differential Effects of Environmental and Genetic Factors on T and B Cell Immune Traits. Cell Rep. 2016, 17, 2474–2487.
- Li, Y.; Oosting, M.; Smeekens, S.P.; Jaeger, M.; Aguirre-Gamboa, R.; Le, K.T.T.; Deelen, P.; Ricaño-Ponce, I.; Schoffelen, T.; Jansen, A.F.M.; et al. A Functional Genomics Approach to Understand Variation in Cytokine Production in Humans. Cell 2016, 167, 1099–1110.e14.
- Quach, H.; Rotival, M.; Pothlichet, J.; Loh, Y.H.E.; Dannemann, M.; Zidane, N.; Laval, G.; Patin, E.; Harmant, C.; Lopez, M.; et al. Genetic Adaptation and Neandertal Admixture Shaped the Immune System of Human Populations. Cell 2016, 167, 643–656.e17.
- Ng, P.C.; Henikoff, S. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003, 31, 3812–3814.
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249.
- Kircher, M.; Witten, D.M.; Jain, P.; O′roak, B.J.; Cooper, G.M.; Shendure, J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 2014, 46, 310–315.
- Barreiro, L.B.; Ben-Ali, M.; Quach, H.; Laval, G.; Patin, E.; Pickrell, J.K.; Bouchier, C.; Tichit, M.; Neyrolles, O.; Gicquel, B.; et al. Evolutionary dynamics of human toll-like receptors and their different contributions to host defense. PLoS Genet. 2009, 5.
- Laayouni, H.; Oosting, M.; Luisi, P.; Ioana, M.; Alonso, S.; Ricano-Ponce, I.; Trynka, G.; Zhernakova, A.; Plantinga, T.S.; Cheng, S.C.; et al. Convergent evolution in European and Rroma populations reveals pressure exerted by plague on Toll-like receptors. Proc. Natl. Acad. Sci. USA 2014, 111, 2668–2673.
- Dannemann, M.; Andrés, A.M.; Kelso, J. Introgression of Neandertal-and Denisovan-like Haplotypes Contributes to Adaptive Variation in Human Toll-like Receptors. Am. J. Hum. Genet. 2016, 98, 22–33.
- Fry, A.E.; Ghansa, A.; Small, K.S.; Palma, A.; Auburn, S.; Diakite, M.; Green, A.; Campino, S.; Teo, Y.Y.; Clark, T.G.; et al. Positive selection of a CD36 nonsense variant in sub-Saharan Africa, but no association with severe malaria phenotypes. Hum. Mol. Genet. 2009, 18, 2683–2692.
- Wang, J.; Li, Y. CD36 tango in cancer: Signaling pathways and functions. Theranostics 2019, 9, 4893–4908.
- Park, Y.M. CD36, a scavenger receptor implicated in atherosclerosis. Exp. Mol. Med. 2014, 46, e99.
- Zhao, L.; Varghese, Z.; Moorhead, J.F.; Chen, Y.; Ruan, X.Z. CD36 and lipid metabolism in the evolution of atherosclerosis. Br. Med. Bull. 2018, 126, 101–112.
- Hughes, A.L.; Nei, M. Pattern of nucleotide substitution at major histocompatibility complex class I loci reveals overdominant selection. Nature 1988, 335, 167–170.
- Hedrick, P.W. Pathogen resistance and genetic variation at MHC loci. Evolution 2002, 56, 1902–1908.
- Prugnolle, F.; Manica, A.; Charpentier, M.; Guégan, J.F.; Guernier, V.; Balloux, F. Pathogen-driven selection and worldwide HLA class I diversity. Curr. Biol. 2005, 15, 1022–1027.
- Walsh, E.C.; Mather, K.A.; Schaffner, S.F.; Farwell, L.; Daly, M.J.; Patterson, N.; Cullen, M.; Carrington, M.; Bugawan, T.L.; Erlich, H.; et al. An integrated haplotype map of the human major histocompatibility complex. Am. J. Hum. Genet. 2003, 73, 580–590.
- De Bakker, P.I.W.; McVean, G.; Sabeti, P.C.; Miretti, M.M.; Green, T.; Marchini, J.; Ke, X.; Monsuur, A.J.; Whittaker, P.; Delgado, M.; et al. A high-resolution HLA and SNP haplotype map for disease association studies in the extended human MHC. Nat. Genet. 2006, 38, 1166–1172.
- Santos, R.F.; Oliveira, L.; Carmo, A.M. Tuning T Cell Activation: The Function of CD6 At the Immunological Synapse and in T Cell Responses. Curr. Drug Targets 2016, 17, 630–639.
- Burgueño-Bucio, E.; Mier-Aguilar, C.A.; Soldevila, G. The multiple faces of CD5. J. Leukoc. Biol. 2019, 105, 891–904.
- Cho, J.-H.; Sprent, J. TCR tuning of T cell subsets. Immunol. Rev. 2018, 283, 129–137.
- Chappell, P.E.; Garner, L.I.; Yan, J.; Metcalfe, C.; Hatherley, D.; Johnson, S.; Robinson, C.V.; Lea, S.M.; Brown, M.H. Structures of CD6 and Its Ligand CD166 Give Insight into Their Interaction. Structure 2015, 23, 1426–1436.
- Bowen, M.A.; Patel, D.D.; Li, X.; Modrell, B.; Malacko, A.R.; Wang, W.C.; Marquardt, H.; Neubauer, M.; Pesando, J.M.; Francke, U.; et al. Cloning, mapping, and characterization of activated leukocyte-cell adhesion molecule (ALCAM), a CD6 ligand. J. Exp. Med. 1995, 181, 2213–2220.
- Levin, T.G.; Powell, A.E.; Davies, P.S.; Silk, A.D.; Dismuke, A.D.; Anderson, E.C.; Swain, J.R.; Wong, M.H. Characterization of the intestinal cancer stem cell marker CD166 in the human and mouse gastrointestinal tract. Gastroenterology 2010, 139, 2072–2082.e5.
- Patel, D.D.; Wee, S.F.; Whichard, L.P.; Bowen, M.A.; Pesando, J.M.; Aruffo, A.; Haynes, B.F. Identification and characterization of a 100-kD ligand for CD6 on human thymic epithelial cells. J. Exp. Med. 1995, 181, 1563–1568.
- Donizy, P.; Zietek, M.; Halon, A.; Leskiewicz, M.; Kozyra, C.; Matkowski, R. Prognostic significance of ALCAM (CD166/MEMD) expression in cutaneous melanoma patients. Diagn. Pathol. 2015, 10, 86.
- Escoda-Ferran, C.; Carrasco, E.; Caballero-Baños, M.; Miró-Julià, C.; Martínez-Florensa, M.; Consuegra-Fernández, M.; Martínez, V.G.; Liu, F.-T.; Lozano, F. Modulation of CD6 function through interaction with Galectin-1 and -3. FEBS Lett. 2014, 588, 2805–2813.
- Enyindah-Asonye, G.; Li, Y.; Ruth, J.H.; Spassov, D.S.; Hebron, K.E.; Zijlstra, A.; Moasser, M.M.; Wang, B.; Singer, N.G.; Cui, H.; et al. CD318 is a ligand for CD6. Proc. Natl. Acad. Sci. USA 2017, 114, E6912–E6921.
- Cayrol, R.; Wosik, K.; Berard, J.L.; Dodelet-Devillers, A.; Ifergan, I.; Kebir, H.; Haqqani, A.S.; Kreymborg, K.; Krug, S.; Moumdjian, R.; et al. Activated leukocyte cell adhesion molecule promotes leukocyte trafficking into the central nervous system. Nat. Immunol. 2008, 9, 137–145.
- Tabbekh, M.; Mokrani-Hammani, M.; Bismuth, G.; Mami-Chouaib, F. T-cell modulatory properties of CD5 and its role in antitumor immune responses. Oncoimmunology 2013, 2, e22841.
- Consuegra-Fernández, M.; Aranda, F.; Simões, I.; Orta, M.; Sarukhan, A.; Lozano, F. CD5 as a target for immune-based therapies. Crit. Rev. Immunol. 2015, 35, 85–115.
- Dennehy, K.M.; Broszeit, R.; Ferris, W.F.; Beyers, A.D. Thymocyte activation induces the association of the proto-oncoprotein c-cbl and ras GTPase-activating protein with CD5. Eur. J. Immunol. 1998, 28, 1617–1625.
- Demydenko, D. c-Cbl mediated ubiquitylation and regulation of cell surface exposure of CD5. Biochem. Biophys. Res. Commun. 2010, 392, 500–504.
- Axtell, R.C.; Xu, L.; Barnum, S.R.; Raman, C. CD5-CK2 Binding/Activation-Deficient Mice Are Resistant to Experimental Autoimmune Encephalomyelitis: Protection Is Associated with Diminished Populations of IL-17-Expressing T Cells in the Central Nervous System. J. Immunol. 2006, 177, 8542–8549.
- Soldevila, G.; Raman, C.; Lozano, F. The immunomodulatory properties of the CD5 lymphocyte receptor in health and disease. Curr. Opin. Immunol. 2011, 23, 310–318.
- Mori, D.; Grégoire, C.; Voisinne, G.; Celis-Gutierrez, J.; Aussel, R.; Girard, L.; Camus, M.; Marcellin, M.; Argenty, J.; Burlet-Schiltz, O.; et al. The T cell CD6 receptor operates a multitask signalosome with opposite functions in T cell activation. J. Exp. Med. 2021, 218, e20201011.
- Gimferrer, I.; Ibáñez, A.; Farnós, M.; Sarrias, M.-R.; Fenutría, R.; Roselló, S.; Zimmermann, P.; David, G.; Vives, J.; Serra-Pagès, C.; et al. The Lymphocyte Receptor CD6 Interacts with Syntenin-1, a Scaffolding Protein Containing PDZ Domains. J. Immunol. 2005, 175, 1406–1414.
- Hassan, N.J.; Simmonds, S.J.; Clarkson, N.G.; Hanrahan, S.; Puklavec, M.J.; Bomb, M.; Barclay, A.N.; Brown, M.H. CD6 Regulates T-Cell Responses through Activation-Dependent Recruitment of the Positive Regulator SLP-76. Mol. Cell. Biol. 2006, 26, 6727–6738.
- Breuning, J.; Brown, M.H. T Cell Costimulation by CD6 Is Dependent on Bivalent Binding of a GADS/SLP-76 Complex. Mol. Cell. Biol. 2017, 37, 71–88.
- Hem, C.D.; Ekornhol, M.; Granum, S.; Sundvold-Gjerstad, V.; Spurkland, A. CD6 and Linker of Activated T Cells are Potential Interaction Partners for T Cell-Specific Adaptor Protein. Scand. J. Immunol. 2017, 85, 104–112.
- Vera, J.; Fenutria, R.; Canadas, O.; Figueras, M.; Mota, R.; Sarrias, M.-R.; Williams, D.L.; Casals, C.; Yelamos, J.; Lozano, F. The CD5 ectodomain interacts with conserved fungal cell wall components and protects from zymosan-induced septic shock-like syndrome. Proc. Natl. Acad. Sci. USA 2009, 106, 1506–1511.
- Sarhan, M.A.; Pham, T.N.Q.; Chen, A.Y.; Michalak, T.I. Hepatitis C Virus Infection of Human T Lymphocytes Is Mediated by CD5. J. Virol. 2012, 86, 3723–3735.
- Mourglia-Ettlin, G.; Miles, S.; Velasco-De-Andrés, M.; Armiger-Borràs, N.; Cucher, M.; Dematteis, S.; Lozano, F. The ectodomains of the lymphocyte scavenger receptors CD5 and CD6 interact with tegumental antigens from Echinococcus granulosus sensu lato and protect mice against secondary cystic echinococcosis. PLoS Negl. Trop. Dis. 2018, 12, e0006891.
- Sarrias, M.-R.; Farnós, M.; Mota, R.; Sánchez-Barbero, F.; Ibáñez, A.; Gimferrer, I.; Vera, J.; Fenutría, R.; Casals, C.; Yélamos, J.; et al. CD6 binds to pathogen-associated molecular patterns and protects from LPS-induced septic shock. Proc. Natl. Acad. Sci. USA 2007, 104, 11724–11729.
- Carrasco, E.; Escoda, C.; Alvarez-Fenrández, C.; Sanchez-Palomino, S.; Carreras, E.; Gatell, J.M.; Gallart, T.; García, F.; Climent, N.; Lozano, F. A role for scavenger-like lymphocyte receptor CD6 in HIV-1 viral infection. AIDS Res. Hum. Retrovir. 2014, 30, A49–A50.
- Velasco-de-Andrés, M.; Català, C.; Casadó-Llombart, S.; Simões, I.; Zaragoza, O.; Carreras, E.; Lozano, F. The lymphocyte scavenger receptor CD5 plays a nonredundant role in fungal infection. Cell. Mol. Immunol. 2021, 18, 498–500.
- Català, C.; Velasco-de Andrés, M.; Casadó-Llombart, S.; Martínez-Florensa, M.; García-Luna, J.; Leyton-Pereira, A.; Aranda, F.; Consuegra-Fernández, M.; Mourglia-Ettlin, G.; Lozano, F. CD6 deficiency confers susceptibility to bacterial sepsis. In Proceedings of the 42nd Congress of Sociedad Española de Inmunología, Madrid, Spain, 24–26 March 2021.
- Lecomte, O.; Bock, J.B.; Birren, B.W.; Vollrath, D.; Parnes, J.R. Molecular linkage of the mouse CD5 and CD6 genes. Immunogenetics 1996, 44, 385–390.
- Padilla, O.; Calvo, J.; Vilà, J.M.; Arman, M.; Gimferrer, I.; Places, L.; Arias, M.T.; Pujana, M.A.; Vives, J.; Lozano, F. Genomic organization of the human CD5 gene. Immunogenetics 2000, 51, 993–1001.
- Jones, N.H.; Clabby, M.L.; Dialynas, D.P.; Huang, H.J.S.; Herzenberg, L.A.; Strominger, J.L. Isolation of complementary DNA clones encoding the human lymphocyte glycoprotein T1/Leu-1. Nature 1986, 323, 346–349.
- Renaudineau, Y.; Hillion, S.; Saraux, A.; Mageed, R.A.; Youinou, P. An alternative exon 1 of the CD5 gene regulates CD5 expression in human B lymphocytes. Blood 2005, 106, 2781–2789.
- Bowen, M.A.; Whitney, G.S.; Neubauer, M.; Starling, G.C.; Palmer, D.; Zhang, J.; Nowak, N.J.; Shows, T.B.; Aruffo, A. Structure and Chromosomal Location of the Human CD6 Gene: Detection of Five Human CD6 Isoforms. J. Immunol. 1997, 158, 1149–1156.
- Bonet, L.; Farnós, M.; Martínez-Florensa, M.; Martínez, V.G.; Lozano, F. Identification of functionally relevant phoshorylatable serine clusters in the cytoplasmic region of the human CD6 lymphocyte surface receptor. FEBS Lett. 2013, 587, 2205–2213.
- Castro, M.A.A.; Oliveira, M.I.; Nunes, R.J.; Fabre, S.; Barbosa, R.; Peixoto, A.; Brown, M.H.; Parnes, J.R.; Bismuth, G.; Moreira, A.; et al. Extracellular Isoforms of CD6 Generated by Alternative Splicing Regulate Targeting of CD6 to the Immunological Synapse. J. Immunol. 2007, 178, 4351–4361.
- Carnero-Montoro, E.; Bonet, L.; Engelken, J.; Bielig, T.; Martínez-Florensa, M.; Lozano, F.; Bosch, E. Evolutionary and functional evidence for positive selection at the human CD5 immune receptor gene. Mol. Biol. Evol. 2012, 29, 811–823.
- Cenit, M.C.; Martínez-Florensa, M.; Consuegra, M.; Bonet, L.; Carnero-Montoro, E.; Armiger, N.; Caballero-Baños, M.; Arias, M.T.; Benitez, D.; Ortego-Centeno, N.; et al. Analysis of ancestral and functionally relevant CD5 variants in systemic lupus erythematosus patients. PLoS ONE 2014, 9, e113090.
- Swaminathan, B.; Cuapio, A.; Alloza, I.; Matesanz, F.; Alcina, A.; García-Barcina, M.; Fedetz, M.; Fernández, Ó.; Lucas, M.; Órpez, T.; et al. Fine Mapping and Functional Analysis of the Multiple Sclerosis Risk Gene CD6. PLoS ONE 2013, 8, e62376.
- Kofler, D.M.; Severson, C.A.; Mousissian, N.; De Jager, P.L.; Hafler, D.A. The CD6 Multiple Sclerosis Susceptibility Allele Is Associated with Alterations in CD4+ T Cell Proliferation. J. Immunol. 2011, 187, 3286–3291.
- Peters, J.E.; Lyons, P.A.; Lee, J.C.; Richard, A.C.; Fortune, M.D.; Newcombe, P.J.; Richardson, S.; Smith, K.G.C. Insight into Genotype-Phenotype Associations through eQTL Mapping in Multiple Cell Types in Health and Immune-Mediated Disease. PLoS Genet. 2016, 12, e1005908.
- Moreno-Estrada, A.; Tang, K.; Sikora, M.; Marqus-Bonet, T.; Casals, F.; Navarro, A.; Calafell, F.; Bertranpetit, J.; Stoneking, M.; Bosch, E. Interrogating 11 fast-evolving genes for signatures of recent positive selection in worldwide human populations. Mol. Biol. Evol. 2009, 26, 2285–2297.
- Eastman, S.W.; Martin-Serrano, J.; Chung, W.; Zang, T.; Bieniasz, P.D. Identification of human VPS37C, a component of endosomal sorting complex required for transport-I important for viral budding. J. Biol. Chem. 2005, 280, 628–636.
- Casadó-Llombart, S.; Velasco-de Andrés, M.; Català, C.; Leyton-Pereira, A.; Suárez, B.; Armiger, N.; Carreras, E.; Esteller, M.; Ricart, E.; Ordás, E.; et al. CD5 and CD6 lymphocyte co-receptor expression and variation impact experimental and clinical inflammatory bowel disease. 2021; submitted.
- Potrony, M.; Carreras, E.; Aranda, F.; Zimmer, L.; Puig-Butille, J.-A.; Tell-Martí, G.; Armiger, N.; Sucker, A.; Giménez-Xavier, P.; Martínez-Florensa, M.; et al. Inherited functional variants of the lymphocyte receptor CD5 influence melanoma survival. Int. J. Cancer 2016, 139, 1297–1302.
- Tamura, K.; Sawada, H.; Izumi, Y.; Fukuda, T.; Utsunomiya, A.; Ikeda, S.; Uike, N.; Tsukada, J.; Kawano, F.; Shibuya, T.; et al. Chronic lymphocytic leukemia (CLL) is rare, but the proportion of T-CLL is high in Japan. Eur. J. Haematol. 2001, 67, 152–157.
- Delgado, J.; Bielig, T.; Bonet, L.; Carnero-Montoro, E.; Puente, X.S.; Colomer, D.; Bosch, E.; Campo, E.; Lozano, F. Impact of the functional CD5 polymorphism A471V on the response of chronic lymphocytic leukaemia to conventional chemotherapy regimens. Br. J. Haematol. 2017, 177, 147–150.
- De Jager, P.L.; Jia, X.; Wang, J.; de Bakker, P.I.W.; Ottoboni, L.; Aggarwal, N.T.; Piccio, L.; Raychaudhuri, S.; Tran, D.; Aubin, C.; et al. Meta-analysis of genome scans and replication identify CD6, IRF8 and TNFRSF1A as new multiple sclerosis susceptibility loci. Nat. Genet. 2009, 41, 776–782.
- Swaminathan, B.; Matesanz, F.; Cavanillas, M.L.; Alloza, I.; Otaegui, D.; Olascoaga, J.; Cénit, M.C.; de las Heras, V.; Barcina, M.G.; Arroyo, R.; et al. Validation of the CD6 and TNFRSF1A loci as risk factors for multiple sclerosis in Spain. J. Neuroimmunol. 2010, 223, 100–103.
- Leppä, V.; Surakka, I.; Tienari, P.J.; Elovaara, I.; Compston, A.; Sawcer, S.; Robertson, N.; de Jager, P.L.; Aubin, C.; Hafler, D.A.; et al. The Genetic Association of Variants in CD6, TNFRSF1A and IRF8 to Multiple Sclerosis: A Multicenter Case-Control Study. PLoS ONE 2011, 6, e18813.
- Sawcer, S.; Hellenthal, G.; Pirinen, M.; Spencer, C.C.A.; Patsopoulos, N.A.; Moutsianas, L.; Dilthey, A.; Su, Z.; Freeman, C.; Hunt, S.E.; et al. Genetic risk and a primary role for cell-mediated immune mechanisms in multiple sclerosis. Nature 2011, 476, 214–219.
- Wagner, M.; Bilinska, M.; Pokryszko-Dragan, A.; Sobczynski, M.; Cyrul, M.; Kusnierczyk, P.; Jasek, M. ALCAM and CD6—Multiple sclerosis risk factors. J. Neuroimmunol. 2014, 276, 98–103.
- Wagner, M.; Wiśniewski, A.; Bilińska, M.; Pokryszko-Dragan, A.; Nowak, I.; Kuśnierczyk, P.; Jasek, M. ALCAM—Novel multiple sclerosis locus interfering with HLA-DRB1*1501. J. Neuroimmunol. 2013, 258, 71–76.
- Zhou, P.; Du, L.F.; Lv, G.Q.; Yu, X.M.; Gu, Y.L.; Li, J.P.; Zhang, C. Functional polymorphisms in CD166/ALCAM gene associated with increased risk for breast cancer in a Chinese population. Breast Cancer Res. Treat. 2011, 128, 527–534.
- Johnson, B.A.; Wang, J.; Taylor, E.M.; Caillier, S.J.; Herbert, J.; Khan, O.A.; Cross, A.H.; De Jager, P.L.; Gourraud, P.A.F.; Cree, B.C.A.; et al. Multiple sclerosis susceptibility alleles in African Americans. Genes Immun. 2010, 11, 343–350.
- Park, T.J.; Kim, H.J.; Kim, J.H.; Bae, J.S.; Cheong, H.S.; Park, B.L.; Shin, H.D. Associations of CD6, TNFRSF1A and IRF8 polymorphisms with risk of inflammatory demyelinating diseases. Neuropathol. Appl. Neurobiol. 2013, 39, 519–530.
- Consuegra-Fernández, M.; Julià, M.; Martínez-Florensa, M.; Aranda, F.; Català, C.; Armiger-Borràs, N.; Arias, M.T.; Santiago, F.; Guilabert, A.; Esteve, A.; et al. Genetic and experimental evidence for the involvement of the CD6 lymphocyte receptor in psoriasis. Cell. Mol. Immunol. 2018, 15, 898–906.
- Zheng, M.; Zhang, L.; Yu, H.; Hu, J.; Cao, Q.; Huang, G.; Huang, Y.; Yuan, G.; Kijlstra, A.; Yang, P. Genetic polymorphisms of cell adhesion molecules in Behçet’s disease in a Chinese Han population. Sci. Rep. 2016, 6, 24974.
- Jostins, L.; Ripke, S.; Weersma, R.K.; Duerr, R.H.; McGovern, D.P.; Hui, K.Y.; Lee, J.C.; Philip Schumm, L.; Sharma, Y.; Anderson, C.A.; et al. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature 2012, 491, 119–124.
- Ellinghaus, D.; Jostins, L.; Spain, S.L.; Cortes, A.; Bethune, J.; Han, B.; Park, Y.R.; Raychaudhuri, S.; Pouget, J.G.; Hübenthal, M.; et al. Analysis of five chronic inflammatory diseases identifies 27 new associations and highlights disease-specific patterns at shared loci. Nat. Genet. 2016, 48, 510–518.
- Darvishi, B.; Boroumandieh, S.; Majidzadeh-A, K.; Salehi, M.; Jafari, F.; Farahmand, L. The role of activated leukocyte cell adhesion molecule (ALCAM) in cancer progression, invasion, metastasis and recurrence: A novel cancer stem cell marker and tumor-specific prognostic marker. Exp. Mol. Pathol. 2020, 115, 104443.
- Eyre, S.; Bowes, J.; Diogo, D.; Lee, A.; Barton, A.; Martin, P.; Zhernakova, A.; Stahl, E.; Viatte, S.; McAllister, K.; et al. High-density genetic mapping identifies new susceptibility loci for rheumatoid arthritis. Nat. Genet. 2012, 44, 1336–1340.