The emergence of three-dimensional human organoids has opened the door for the development of patient-derived cancer organoid (PDO) models, which closely recapitulate parental tumor tissue.
- organoid
- stem cell
- cancer
- glioblastoma
1. Introduction
The recent development of organoids, defined as in vitro three-dimensional models of human tissues, has allowed for more accurate recapitulation of parent organs [6,7,8,9,10,11,12,13,14,15,16,17][1][2][3][4][5][6][7][8][9][10][11][12]. The available literature describing patient-derived cancer organoids suggests that organoid cancer models consistently reflect in vivo tumor genotypes, phenotypes, intertumor heterogeneity, and cell–cell and cell–stroma interactions and are less resource and time intensive compared to existing mouse models [23,24,25,26,27,28,29,30,31,32][13][14][15][16][17][18][19][20][21][22]. Organoid cancer models may mitigate challenges presented by existing cancer models and augment the implementation of precision oncology by providing personalized platforms to study each patient’s tumor.
2. History of Organoid Development
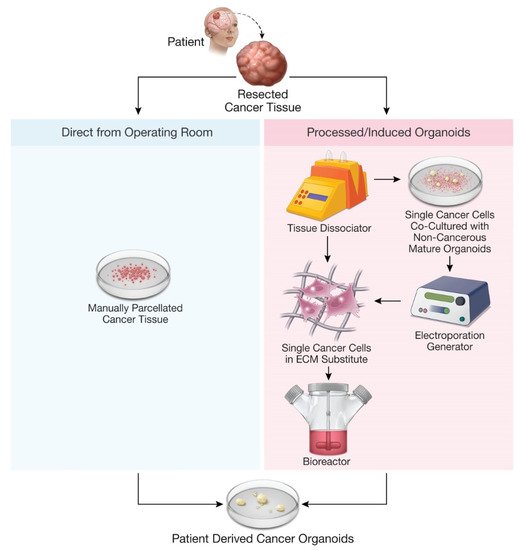
3. Current Models of Patient-Derived Cancer Glioma Organoids
| Cancer Type | Source of Organoids | Culture Technique | Endpoint of Study | Resemblance to Parent Tumor | References |
|---|
| Glioblastoma | Patient-derived glioma stem cells | 1. Co-culture of GSCs and iPSCs 2. Supplementing GSCs with normal cerebral organoids 3. Fusion of GSC spheres with normal brain organoids |
Three techniques to model GSC invasion in normal brain organoids and creation of GBM organoids | No direct comparison to primary tissue could be made. | Goranci-Buzhala et al. (2020) [83] | Goranci-Buzhala et al. (2020) [54] |
| Patient-derived glioblastoma and non-glioblastoma stem cells | Matrigel + culture shaking in NBM complete media | Spatial distribution of GBM replicated in organoids derived from glioblastoma and non-glioblastoma stem cells | Orthotopically implanted PDOs were diffuse and infiltrative, histologically resembling the parent tumor. | Hubert et al. (2016) [57] | Hubert et al. (2016) [47] | |
| Human embryonic stem cells | Normal cerebral organoids co-cultured with patient-derived tumor cells or oncogene introduction through electroporation | Normal human-derived cerebral organoids a vector for glioblastoma organoid modeling | Engineered PDOs displayed a mesenchymal phenotype consistent with patient-derived GBMs on transcriptomic analysis. | Ogawa et al. (2018) [56] | Ogawa et al. (2018) [46] | |
| Human glioblastoma tissue | Patient tissue parcellated and cultured without an extracellular matrix | Glioblastoma organoid protocol development from primary tissue samples with minimum processing | PDOs had cellular and nuclear atypia, abundant mitotic figures, and pleomorphic nuclei consistent with high-grade parent tumors on histologic analysis. | Jacob et al. (2020) [62] | Jacob et al. (2020) [52] | |
| Glioblastoma spheroids infiltrating cerebral organoids | Co-culture of mouse early (6-day-old) cerebral organoids with glioblastoma spheroids created from glioblastoma stem cell culture | Demonstration of hybrid glioblastoma organoid modeling | Co-cultured organoids displayed core infiltration and expression of GBM stem cell markers NESTIN and SOX2. | Da Silva et al. (2018) [82] | Da Silva et al. (2018) [53] | |
| Patient-derived glioma stem cells | Co-culture of cerebral organoids with transduced GSCs on NBM | Single-cell RNA sequencing comparison of four patient-derived glioblastoma models | Organoids displayed microscopic invasion and single-cell heterogeneity consistent with parent tumors. | Pine et al. (2020) [85] | Pine et al. (2020) [57] | |
| Central nervous system primitive neuroectodermal-like and glioblastoma-like tumor | Human embryonic stem cells | Neoplastic cerebral organoids (Neo-COR); combination of plasmids introduced into normal cerebral organoids through electroporation before te organoids embedded in Matrigel | Demonstration of brain tumorigenesis through introduction of oncogenic mutations in normal cerebral organoids through transposon and CRISPR/Cas9 mutagenesis | GBM-like Neo-COR displayed upregulation of GBM genes on transcriptomic analysis. | Bian et al. (2018) [68] | Bian et al. (2018) [55] |
References
- Seidel, S.; Garvalov, B.K.; Acker, T. Isolation and culture of primary glioblastoma cells from human tumor specimens. Methods Mol. Biol. 2014.
- Aboulkheyr Es, H.; Montazeri, L.; Aref, A.R.; Vosough, M.; Baharvand, H. Personalized Cancer Medicine: An Organoid Approach. Trends Biotechnol. 2018.
- Fan, H.; Demirci, U.; Chen, P. Emerging organoid models: Leaping forward in cancer research. J. Hematol. Oncol. 2019.
- Walker, K.; Hjelmeland, A. Method for Efficient Transduction of Cancer Stem Cells. J. Cancer Stem Cell Res. 2014.
- Feist, P.E.; Sidoli, S.; Liu, X.; Schroll, M.M.; Rahmy, S.; Fujiwara, R.; Garcia, B.A.; Hummon, A.B. Multicellular Tumor Spheroids Combined with Mass Spectrometric Histone Analysis to Evaluate Epigenetic Drugs. Anal. Chem. 2017, 89, 2773–2781.
- Shergalis, A.; Bankhead, A.; Luesakul, U.; Muangsin, N.; Neamati, N. Current challenges and opportunities in treating glioblastomas. Pharmacol. Rev. 2018.
- Dirkse, A.; Golebiewska, A.; Buder, T.; Nazarov, P.V.; Muller, A.; Poovathingal, S.; Brons, N.H.C.; Leite, S.; Sauvageot, N.; Sarkisjan, D.; et al. Stem cell-associated heterogeneity in Glioblastoma results from intrinsic tumor plasticity shaped by the microenvironment. Nat. Commun. 2019, 10, 1–16.
- Zahalka, A.H.; Frenette, P.S. Nerves in cancer. Nat. Rev. Cancer 2020.
- Quail, D.F.; Joyce, J.A. The Microenvironmental Landscape of Brain Tumors. Cancer Cell. 2017, 31, 326–341.
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016.
- Claus, E.B.; Walsh, K.M.; Wiencke, J.K.; Molinaro, A.M.; Wiemels, J.L.; Schildkraut, J.M.; Bondy, M.L.; Berger, M.; Jenkins, R.; Wrensch, M. Survival and low-grade glioma: The emergence of genetic information. Neurosurg. Focus 2015.
- Zhang, L.; Liu, Y.; Wang, M.; Wu, Z.; Li, N.; Zhang, J.; Yang, C. EZH2−, CHD4−, and IDH-linked epigenetic perturbation and its association with survival in glioma patients. J. Mol. Cell Biol. 2017.
- Shi, R.; Radulovich, N.; Ng, C.; Liu, N.; Notsuda, H.; Cabanero, M.; Martins-Filho, S.N.; Raghavan, V.; Li, Q.; Mer, A.S.; et al. Organoid cultures as preclinical models of non-small cell lung cancer. Clin. Cancer Res. 2020, 26, 1162–1174.
- Boj, S.F.; Hwang, C., II; Baker, L.A.; Chio, I.I.C.; Engle, D.D.; Corbo, V.; Jager, M.; Ponz-Sarvise, M.; Tiriac, H.; Spector, M.S.; et al. Organoid models of human and mouse ductal pancreatic cancer. Cell 2015, 160, 324–338.
- Broutier, L.; Mastrogiovanni, G.; Verstegen, M.M.A.; Francies, H.E.; Gavarró, L.M.; Bradshaw, C.R.; Allen, G.E.; Arnes-Benito, R.; Sidorova, O.; Gaspersz, M.P.; et al. Human primary liver cancer-derived organoid cultures for disease modeling and drug screening. Nat. Med. 2017, 23, 1424.
- Gao, D.; Vela, I.; Sboner, A.; Iaquinta, P.J.; Karthaus, W.R.; Gopalan, A.; Dowling, C.; Wanjala, J.N.; Undvall, E.A.; Arora, V.K.; et al. Organoid cultures derived from patients with advanced prostate cancer. Cell 2014, 159, 176–187.
- Kopper, O.; de Witte, C.J.; Lõhmussaar, K.; Valle-Inclan, J.E.; Hami, N.; Kester, L.; Balgobind, A.V.; Korving, J.; Proost, N.; Begthel, H.; et al. An organoid platform for ovarian cancer captures intra- and interpatient heterogeneity. Nat. Med. 2019, 25, 838–849.
- Sachs, N.; de Ligt, J.; Kopper, O.; Gogola, E.; Bounova, G.; Weeber, F.; Balgobind, A.V.; Wind, K.; Gracanin, A.; Begthel, H.; et al. A Living Biobank of Breast Cancer Organoids Captures Disease Heterogeneity. Cell 2018, 172, 373–386.
- Yan, H.H.N.; Siu, H.C.; Law, S.; Ho, S.L.; Yue, S.S.K.; Tsui, W.Y.; Chan, D.; Chan, A.S.; Ma, S.; Lam, K.O.; et al. A Comprehensive Human Gastric Cancer Organoid Biobank Captures Tumor Subtype Heterogeneity and Enables Therapeutic Screening. Cell Stem Cell 2018, 23, 882–897.
- Van De Wetering, M.; Francies, H.E.; Francis, J.M.; Bounova, G.; Iorio, F.; Pronk, A.; Van Houdt, W.; Van Gorp, J.; Taylor-Weiner, A.; Kester, L.; et al. Prospective derivation of a living organoid biobank of colorectal cancer patients. Cell 2015, 161, 933–945.
- Driehuis, E.; Van Hoeck, A.; Moore, K.; Kolders, S.; Francies, H.E.; Gulersonmez, M.C.; Stigter, E.C.A.; Burgering, B.; Geurts, V.; Gracanin, A.; et al. Pancreatic cancer organoids recapitulate disease and allow personalized drug screening. Proc. Natl. Acad. Sci. USA 2019, 116, 26580–26590.
- Dutta, D.; Heo, I.; Clevers, H. Disease Modeling in Stem Cell-Derived 3D Organoid Systems. Trends Mol. Med. 2017.
- Bartfeld, S.; Clevers, H. Stem cell-derived organoids and their application for medical research and patient treatment. J. Mol. Med. 2017.
- Fatehullah, A.; Tan, S.H.; Barker, N. Organoids as an in vitro model of human development and disease. Nat. Cell Biol. 2016.
- Dzobo, K.; Rowe, A.; Senthebane, D.A.; Almazyadi, M.A.M.; Patten, V.; Parker, M.I. Three-Dimensional Organoids in Cancer Research: The Search for the Holy Grail of Preclinical Cancer Modeling. Omics J. Integr. Biol. 2018.
- Sato, T.; Vries, R.G.; Snippert, H.J.; Van De Wetering, M.; Barker, N.; Stange, D.E.; Van Es, J.H.; Abo, A.; Kujala, P.; Peters, P.J.; et al. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature 2009, 459, 262–265.
- Lancaster, M.A.; Knoblich, J.A. Generation of cerebral organoids from human pluripotent stem cells. Nat. Protoc. 2014.
- Forster, R.; Chiba, K.; Schaeffer, L.; Regalado, S.G.; Lai, C.S.; Gao, Q.; Kiani, S.; Farin, H.F.; Clevers, H.; Cost, G.J.; et al. Human intestinal tissue with adult stem cell properties derived from pluripotent stem cells. Stem Cell Rep. 2014.
- Ren, W.; Lewandowski, B.C.; Watson, J.; Aihara, E.; Iwatsuki, K.; Bachmanov, A.A.; Margolskee, R.F.; Jiang, P. Single Lgr5- or Lgr6-expressing taste stem/progenitor cells generate taste bud cells ex vivo. Proc. Natl. Acad. Sci. USA 2014, 111, 16401–16406.
- Eiraku, M.; Takata, N.; Ishibashi, H.; Kawada, M.; Sakakura, E.; Okuda, S.; Sekiguchi, K.; Adachi, T.; Sasai, Y. Self-organizing optic-cup morphogenesis in three-dimensional culture. Nature 2011, 472, 51–56.
- Takasato, M.; Er, P.X.; Chiu, H.S.; Little, M.H. Generation of kidney organoids from human pluripotent stem cells. Nat. Protoc. 2016.
- Wimmer, R.A.; Leopoldi, A.; Aichinger, M.; Kerjaschki, D.; Penninger, J.M. Generation of blood vessel organoids from human pluripotent stem cells. Nat. Protoc. 2019.
- Miller, A.J.; Dye, B.R.; Ferrer-Torres, D.; Hill, D.R.; Overeem, A.W.; Shea, L.D.; Spence, J.R. Generation of lung organoids from human pluripotent stem cells in vitro. Nat. Protoc. 2019.
- Bagley, J.A.; Reumann, D.; Bian, S.; Lévi-Strauss, J.; Knoblich, J.A. Fused cerebral organoids model interactions between brain regions. Nat. Methods 2017.
- Lancaster, M.A.; Renner, M.; Martin, C.A.; Wenzel, D.; Bicknell, L.S.; Hurles, M.E.; Homfray, T.; Penninger, J.M.; Jackson, A.P.; Knoblich, J.A. Cerebral organoids model human brain development and microcephaly. Nature 2013, 501, 373–379.
- Roerink, S.F.; Sasaki, N.; Lee-Six, H.; Young, M.D.; Alexandrov, L.B.; Behjati, S.; Mitchell, T.J.; Grossmann, S.; Lightfoot, H.; Egan, D.A.; et al. Intra-tumour diversification in colorectal cancer at the single-cell level. Nature 2018, 556, 457–462.
- Yucer, N.; Holzapfel, M.; Jenkins Vogel, T.; Lenaeus, L.; Ornelas, L.; Laury, A.; Sareen, D.; Barrett, R.; Karlan, B.Y.; Svendsen, C.N. Directed Differentiation of Human Induced Pluripotent Stem Cells into Fallopian Tube Epithelium. Sci. Rep. 2017.
- Sato, T.; Stange, D.E.; Ferrante, M.; Vries, R.G.J.; Van Es, J.H.; Van Den Brink, S.; Van Houdt, W.J.; Pronk, A.; Van Gorp, J.; Siersema, P.D.; et al. Long-term expansion of epithelial organoids from human colon, adenoma, adenocarcinoma, and Barrett’s epithelium. Gastroenterology 2011.
- Drost, J.; Van Jaarsveld, R.H.; Ponsioen, B.; Zimberlin, C.; Van Boxtel, R.; Buijs, A.; Sachs, N.; Overmeer, R.M.; Offerhaus, G.J.; Begthel, H.; et al. Sequential cancer mutations in cultured human intestinal stem cells. Nature 2015, 521, 43–47.
- Matano, M.; Date, S.; Shimokawa, M.; Takano, A.; Fujii, M.; Ohta, Y.; Watanabe, T.; Kanai, T.; Sato, T. Modeling colorectal cancer using CRISPR-Cas9-mediated engineering of human intestinal organoids. Nat. Med. 2015.
- Verissimo, C.S.; Overmeer, R.M.; Ponsioen, B.; Drost, J.; Mertens, S.; Verlaan-Klink, I.; Van Gerwen, B.; Van Der Ven, M.; Van De Wetering, M.; Egan, D.A.; et al. Targeting mutant RAS in patient-derived colorectal cancer organoids by combinatorial drug screening. eLife 2016.
- Weeber, F.; Van De Wetering, M.; Hoogstraat, M.; Dijkstra, K.K.; Krijgsman, O.; Kuilman, T.; Gadellaa-Van Hooijdonk, C.G.M.; Van Der Velden, D.L.; Peeper, D.S.; Cuppen, E.P.J.G.; et al. Preserved genetic diversity in organoids cultured from biopsies of human colorectal cancer metastases. Proc. Natl. Acad. Sci. USA 2015.
- Corrò, C.; Novellasdemunt, L.; Li, V.S.W. A brief history of organoids. Am. J. Physiol. Cell Physiol. 2020, 319, C151–C165.
- Drost, J.; Clevers, H. Translational applications of adult stem cell-derived organoids. Development 2017.
- Li, X.; Francies, H.E.; Secrier, M.; Perner, J.; Miremadi, A.; Galeano-Dalmau, N.; Barendt, W.J.; Letchford, L.; Leyden, G.M.; Goffin, E.K.; et al. Organoid cultures recapitulate esophageal adenocarcinoma heterogeneity providing a model for clonality studies and precision therapeutics. Nat. Commun. 2018.
- Ogawa, J.; Pao, G.M.; Shokhirev, M.N.; Verma, I.M. Glioblastoma Model Using Human Cerebral Organoids. Cell Rep. 2018.
- Hubert, C.G.; Rivera, M.; Spangler, L.C.; Wu, Q.; Mack, S.C.; Prager, B.C.; Couce, M.; McLendon, R.E.; Sloan, A.E.; Rich, J.N. A three-dimensional organoid culture system derived from human glioblastomas recapitulates the hypoxic gradients and cancer stem cell heterogeneity of tumors found in vivo. Cancer Res. 2016.
- Jin, M.Z.; Han, R.R.; Qiu, G.Z.; Ju, X.C.; Lou, G.; Jin, W.L. Organoids: An intermediate modeling platform in precision oncology. Cancer Lett. 2018.
- Lee, S.H.; Hu, W.; Matulay, J.T.; Silva, M.V.; Owczarek, T.B.; Kim, K.; Chua, C.W.; Barlow, L.M.J.; Kandoth, C.; Williams, A.B.; et al. Tumor Evolution and Drug Response in Patient-Derived Organoid Models of Bladder Cancer. Cell 2018, 173, 515–528.
- Karthaus, W.R.; Iaquinta, P.J.; Drost, J.; Gracanin, A.; Van Boxtel, R.; Wongvipat, J.; Dowling, C.M.; Gao, D.; Begthel, H.; Sachs, N.; et al. Identification of multipotent luminal progenitor cells in human prostate organoid cultures. Cell 2014.
- Jacob, F.; Ming, G.L.; Song, H. Generation and biobanking of patient-derived glioblastoma organoids and their application in CAR T cell testing. Nat. Protoc. 2020.
- Jacob, F.; Salinas, R.D.; Zhang, D.Y.; Nguyen, P.T.T.; Schnoll, J.G.; Wong, S.Z.H.; Thokala, R.; Sheikh, S.; Saxena, D.; Prokop, S.; et al. A Patient-Derived Glioblastoma Organoid Model and Biobank Recapitulates Inter- and Intra-tumoral Heterogeneity. Cell 2020.
- Da Silva, B.; Mathew, R.K.; Polson, E.S.; Williams, J.; Wurdak, H. Spontaneous Glioblastoma Spheroid Infiltration of Early-Stage Cerebral Organoids Models Brain Tumor Invasion. SLAS Discov. 2018.
- Goranci-Buzhala, G.; Mariappan, A.; Gabriel, E.; Ramani, A.; Ricci-Vitiani, L.; Buccarelli, M.; D’Alessandris, Q.G.; Pallini, R.; Gopalakrishnan, J. Rapid and Efficient Invasion Assay of Glioblastoma in Human Brain Organoids. Cell Rep. 2020.
- Bian, S.; Repic, M.; Guo, Z.; Kavirayani, A.; Burkard, T.; Bagley, J.A.; Krauditsch, C.; Knoblich, J.A. Genetically engineered cerebral organoids model brain tumor formation. Nat. Methods 2018.
- Degl’Innocenti, A.; di Leo, N.; Ciofani, G. Genetic Hallmarks and Heterogeneity of Glioblastoma in the Single-Cell Omics Era. Adv. Ther. 2020.
- Pine, A.R.; Cirigliano, S.M.; Nicholson, J.G.; Hu, Y.; Linkous, A.; Miyaguchi, K.; Edwards, L.; Singhania, R.; Schwartz, T.H.; Ramakrishna, R.; et al. Tumor microenvironment is critical for the maintenance of cellular states found in primary glioblastomas. Cancer Discov. 2020.
- Pistollato, F.; Abbadi, S.; Rampazzo, E.; Persano, L.; Della Puppa, A.; Frasson, C.; Sarto, E.; Scienza, R.; D’Avella, D.; Basso, G. Intratumoral hypoxic gradient drives stem cells distribution and MGMT expression in glioblastoma. Stem Cells 2010.
- Li, Z.; Bao, S.; Wu, Q.; Wang, H.; Eyler, C.; Sathornsumetee, S.; Shi, Q.; Cao, Y.; Lathia, J.; McLendon, R.E.; et al. Hypoxia-Inducible Factors Regulate Tumorigenic Capacity of Glioma Stem Cells. Cancer Cell 2009.
- Calabrese, C.; Poppleton, H.; Kocak, M.; Hogg, T.L.; Fuller, C.; Hamner, B.; Oh, E.Y.; Gaber, M.W.; Finklestein, D.; Allen, M.; et al. A Perivascular Niche for Brain Tumor Stem Cells. Cancer Cell 2007.
- Seidel, S.; Garvalov, B.K.; Wirta, V.; Von Stechow, L.; Schänzer, A.; Meletis, K.; Wolter, M.; Sommerlad, D.; Henze, A.T.; Nistér, M.; et al. A hypoxic niche regulates glioblastoma stem cells through hypoxia inducible factor 2α. Brain 2010.
- Furnari, F.B.; Cloughesy, T.F.; Cavenee, W.K.; Mischel, P.S. Heterogeneity of epidermal growth factor receptor signalling networks in glioblastoma. Nat. Rev. Cancer 2015.
- Brandes, A.A.; Franceschi, E.; Tosoni, A.; Blatt, V.; Pession, A.; Tallini, G.; Bertorelle, R.; Bartolini, S.; Calbucci, F.; Andreoli, A.; et al. MGMT promoter methylation status can predict the incidence and outcome of pseudoprogression after concomitant radiochemotherapy in newly diagnosed glioblastoma patients. J. Clin. Oncol. 2008.
- Golebiewska, A.; Hau, A.C.; Oudin, A.; Stieber, D.; Yabo, Y.A.; Baus, V.; Barthelemy, V.; Klein, E.; Bougnaud, S.; Keunen, O.; et al. Patient-derived organoids and orthotopic xenografts of primary and recurrent gliomas represent relevant patient avatars for precision oncology. Acta Neuropathol. 2020.
- Zhang, B.; Shen, R.; Cheng, S.; Feng, L. Immune microenvironments differ in immune characteristics and outcome of glioblastoma multiforme. Cancer Med. 2019.
- Neal, J.T.; Li, X.; Zhu, J.; Giangarra, V.; Grzeskowiak, C.L.; Ju, J.; Liu, I.H.; Chiou, S.H.; Salahudeen, A.A.; Smith, A.R.; et al. Organoid Modeling of the Tumor Immune Microenvironment. Cell 2018, 175, 1972–1988.
- Venkatesh, H.S.; Morishita, W.; Geraghty, A.C.; Silverbush, D.; Gillespie, S.M.; Arzt, M.; Tam, L.T.; Espenel, C.; Ponnuswami, A.; Ni, L.; et al. Electrical and synaptic integration of glioma into neural circuits. Nature 2019.
- Hambardzumyan, D.; Bergers, G. Glioblastoma: Defining Tumor Niches. Trends Cancer 2015.