The decreasing sequencing cost and the rapid expansion of our knowledge of human disease genes has fueled a significant increase in the uptake and utilization of clinical genomic testing. It is essential that all healthcare providers understand the basic concepts of genetic testing and how to properly utilize this testing for each patient. There are several factors that need to be considered when choosing the appropriate testing strategy including and their utilization based on different clinical scenarios, test characteristics such as the types of genetic variation identified by each test, turnaround time, and diagnostic yield for different clinical indications. Effective application of this knowledge will aid healthcare providers in utilizing the most appropriate, fastest, and most cost-effective genetic test for their patients, thereby increasing the likelihood of a timely diagnosis and reducing the financial burden on the healthcare system by eliminating unnecessary and redundant testing. This entry is not comprehensive, but will provide the basic knowledge to understand the differences between various testing strategies and their limitations.
- genetic testing
- diagnostics
- personalized medicine
- genetics
- genomics
- next generation sequencing
- pediatrics
- whole exome sequencing
- whole genome sequencing
1. Genomic Testing Strategies
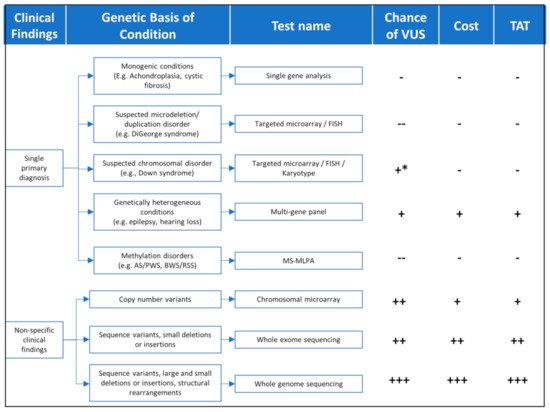
Figure 1.
Table 1.
| Test Name | Variants Identified | When to Use | Limitations | Diagnostic Yield | Turnaround Time ** | References |
|---|
| Chromosomal microarray | Large chromosomal deletion/duplications High resolution (up to 25 kb) |
MCA, ID or DD, and ASD | Cannot identify balanced rearrangements Cannot identify sequence variants or small copy number variants ≤1–10 kb |
15–20% | 2–6 weeks+ | [1] |
| Karyotype | Large chromosomal deletion/duplications Balanced rearrangements (translocations/inversions) Low resolution (3–5 Mb) |
Determine recurrence risks for aneuploidies (e.g., Down syndrome) FHx of Balanced translocations Hx of multiple miscarriages |
Low resolution Cannot identify sequence variants or copy number variants <3 MB |
~3% | 1–2 weeks+ | [1] |
| Single gene sequencing and/or del/dup analysis | Sequence variants, small deletions (<50 bp), or insertions | Phenotype is specific for a single monogenic disorder | Very few conditions are monogenic | Up to 100% depending on clinical ascertainment | 1–4 weeks+ | |
| Panels (Preset or customizable) |
Sequence variants, small deletions (<50 bp *), or insertions * Larger copy number variants can be identified by some laboratories |
Clearly defined phenotype Short differential diagnosis list |
Not suitable if no primary phenotype Not suitable if differential diagnosis is long |
Variable between condition/phenotype | 2–6 weeks+ | [2][3][4][5] |
| Whole exome sequencing | Sequence variants, small deletions (<50 bp *), or insertions (* Larger copy number variants can be identified by some laboratories) |
Unclear/large differential diagnosis Non-specific clinical findings Specific test unavailable |
Relatively expensive Long turnaround time High chance of VUS Secondary findings Incidental findings |
31–53% 8–70% 16–58% |
5–12 weeks+ | [6][7][8] |
| Whole genome sequencing | Sequence variants, small deletions, or insertions Structural rearrangements, including balanced translocations, inversions, and deletion/duplications (large and small) |
Unclear/large differential diagnosis Non-specific clinical findings Specific test unavailable |
Very expensive Long turnaround time High chance of VUS Limited understanding of non-coding variants Secondary findings Incidental findings |
8–70% 36–86% |
4–16 weeks+ | [7][8] |
| Triplet repeat analysis | Expansion of triplet repeats | Triplet repeat expansions, e.g., Fragile X, myotonic dystrophy, spinocerebellar ataxia | Requires knowledge of conditions caused by triplet repeat expansions | Varies by clinical ascertainment | 2–4 weeks+ | [9][10][11] |
| Methylation analysis | Imprinting/methylation disorders/ Uniparental disomy |
BWS/RSS, PWS/AS | Requires knowledge of conditions caused by imprinting defects | Variable between conditions | 2–4 weeks+ | [12][13][14][15] |
MCA = multiple congenital anomalies; ID = intellectual disability; DD = developmental delay; ASD = autism spectrum disorder; BWS = Beckwith–Wiedemann syndrome; RSS = Russell–Silver syndrome; PWS = Prader–Willi syndrome; AS = Angelman syndrome; VUS = variant of uncertain significance; FHx = Family history; Hx = History; ** = Turnaround times vary by lab; times stated taken from a survey of labs obtained from concertgenetics.com.
FGFR3
COL1A1
COL1A2
FMRP
1.1. Chromosomal Microarray
[1]
[16]
1.2. Whole Exome Sequencing
[6]
[6]
[7]
[19]
[20]
1.3. Targeted Gene Panels
[24]
1.4. Customizable Gene Panels
1.5. Single Gene Analysis
FGFR3
[29]
CFTR
[30]
CFTR
1.6. Triplet Repeat Disorders
FMR1
[11]
FMRP
1.7. Methylation Disorders
[32]
UBE3A
UBE3A
[15]
1.8. Whole Genome Sequencing
[33]
[34]
[35]
2.1. Interpreting VUS
USH2A
[39]
2.2. Negative or Inconclusive Report
2.3. Diagnostic Yield
EYA1
SIX1
SIX5
[43]
2.4. Insurance Coverage
References
- David T. Miller; Margaret P. Adam; Swaroop Aradhya; Leslie G. Biesecker; Arthur R. Brothman; Nigel P. Carter; Deanna M. Church; John A. Crolla; Evan E. Eichler; Charles J. Epstein; et al.W. Andrew FaucettLars FeukJan M. FriedmanAda HamoshLaird JacksonErin B. KaminskyKlaas KokIan D. KrantzRobert M. KuhnCharles LeeJames M. OstellCarla RosenbergStephen W. SchererNancy B. SpinnerDimitri J. StavropoulosJames H. TepperbergErik C. ThorlandJoris R. VermeeschDarrel J. WaggonerMichael S. WatsonChrista Lese MartinDavid H. Ledbetter Consensus Statement: Chromosomal Microarray Is a First-Tier Clinical Diagnostic Test for Individuals with Developmental Disabilities or Congenital Anomalies. The American Journal of Human Genetics 2010, 86, 749-764, 10.1016/j.ajhg.2010.04.006.
- Basil M. Fathalla; Ali Alsarhan; Samina Afzal; Maha El Naofal; Ahmad Abou Tayoun; The genomic landscape of pediatric rheumatology disorders in the Middle East. Human Mutation 2021, 42, e1-e14, 10.1002/humu.24165.
- Scarffe, Leslie; Alsubhi, Sarah; Accogli, Andrea; Srour, Myriam; The diagnostic yield of next generation sequencing panels for epilepsy in clinical practice (2229). Neurology 2020, 94(15), 2229.
- Jorune Balciuniene; Elizabeth T. DeChene; Gozde Akgumus; Edward J. Romasko; Kajia Cao; Holly A. Dubbs; Surabhi Mulchandani; Nancy B. Spinner; Laura K. Conlin; Eric D. Marsh; et al.Ethan GoldbergIngo HelbigMahdi SarmadyAhmad Abou Tayoun Use of a Dynamic Genetic Testing Approach for Childhood-Onset Epilepsy. JAMA Network Open 2019, 2, e192129, 10.1001/jamanetworkopen.2019.2129.
- Heather Pekeles; Andrea Accogli; Nassima Boudrahem-Addour; Laura Russell; Fabienne Parente; Myriam Srour; Diagnostic Yield of Intellectual Disability Gene Panels. Pediatric Neurology 2019, 92, 32-36, 10.1016/j.pediatrneurol.2018.11.005.
- Siddharth Srivastava; the NDD Exome Scoping Review Work Group; Jamie A. Love-Nichols; Kira A. Dies; David H. Ledbetter; Christa L. Martin; Wendy K. Chung Md; Helen V. Firth; Thomas Frazier; Robin L. Hansen; et al.Lisa ProckHan BrunnerNy HoangStephen W. SchererMustafa SahinDavid T. Miller Md Meta-analysis and multidisciplinary consensus statement: exome sequencing is a first-tier clinical diagnostic test for individuals with neurodevelopmental disorders. Genetics in Medicine 2019, 21, 2413-2421, 10.1038/s41436-019-0554-6.
- Caroline F. Wright; David R. Fitzpatrick; Helen V. Firth; Paediatric genomics: diagnosing rare disease in children. Nature Reviews Genetics 2018, 19, 253-268, 10.1038/nrg.2017.116.
- John S Mattick; Marcel Dinger; Nicole Schonrock; Mark Cowley; Whole genome sequencing provides better diagnostic yield and future value than whole exome sequencing. Medical Journal of Australia 2018, 209, 197-199, 10.5694/mja17.01176.
- Myotonic Dystrophy Type 1 . GeneReviews®. Retrieved 2021-6-8
- Spinocerebellar Ataxia Type 1 . GeneReviews®. Retrieved 2021-6-8
- FMR1 disorders . GeneReviews®. Retrieved 2021-6-8
- Beckwith-Wiedemann Syndrome . GeneReviews® . Retrieved 2021-6-8
- Silver-Russell Syndrome . GeneReviews®. Retrieved 2021-6-8
- Prader-Willi Syndrome . GeneReviews®. Retrieved 2021-6-8
- Angelman Syndrome . GeneReviews®. Retrieved 2021-6-8
- Fabio Coppedè; Risk factors for Down syndrome. Archives of Toxicology 2016, 90, 2917-2929, 10.1007/s00204-016-1843-3.
- Ye K; Hall G; Ning Z; Structural Variation Detection from Next Generation Sequencing. Journal of Next Generation Sequencing & Applications 2015, 01, 1 - 6, 10.4172/2469-9853.s1-007.
- José Marcos Moreno-Cabrera; Jesús Del Valle; Elisabeth Castellanos; Lidia Feliubadaló; Marta Pineda; Joan Brunet; Eduard Serra; Gabriel Capellà; Conxi Lázaro; Bernat Gel; et al. Evaluation of CNV detection tools for NGS panel data in genetic diagnostics. European Journal of Human Genetics 2020, 28, 1645-1655, 10.1038/s41431-020-0675-z.
- Sue Richards; Nazneen Aziz; Sherri Bale; David Bick; Soma Das; Julie Gastier-Foster; Wayne W. Grody Md; Madhuri Hegde; Elaine Lyon; Elaine Spector; et al.Karl VoelkerdingHeidi L. Rehm Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in Medicine 2015, 17, 405-423, 10.1038/gim.2015.30.
- Sarah S. Kalia; Kathy Adelman; Sherri J. Bale; Wendy K. Chung; Christine Eng; James P. Evans; Gail E. Herman; Sophia B. Hufnagel; Teri E. Klein; Bruce R. Korf; et al.Kent D. McKelveyKelly E. OrmondC. Sue RichardsChristopher N. VlangosMichael WatsonChrista L. MartinDavid T. Milleron behalf of the ACMG Secondary Findings Maintenance Working Group Recommendations for reporting of secondary findings in clinical exome and genome sequencing, 2016 update (ACMG SF v2.0): a policy statement of the American College of Medical Genetics and Genomics. Genetics in Medicine 2016, 19, 249-255, 10.1038/gim.2016.190.
- Natasha T. Strande; Erin Rooney Riggs; Adam H. Buchanan; Ozge Ceyhan-Birsoy; Marina DiStefano; Selina S. Dwight; Jenny Goldstein; Rajarshi Ghosh; Bryce A. Seifert; Tam P. Sneddon; et al.Mathew WrightLaura V. MilkoJ. Michael CherryMonica A. GiovanniMichael F. MurrayJulianne M. O’DanielErin M. RamosAvni B. SantaniAlan F. ScottSharon E. PlonHeidi L. RehmChrista L. MartinJonathan S. Berg Evaluating the Clinical Validity of Gene-Disease Associations: An Evidence-Based Framework Developed by the Clinical Genome Resource. The American Journal of Human Genetics 2017, 100, 895-906, 10.1016/j.ajhg.2017.04.015.
- Ahmad N. Abou Tayoun; Saeed H. Al Turki; Andrea M. Oza; Mark J. Bowser; Amy L. Hernandez; Birgit H. Funke; Heidi L. Rehm; Sami S. Amr; Improving hearing loss gene testing: a systematic review of gene evidence toward more efficient next-generation sequencing–based diagnostic testing and interpretation. Genetics in Medicine 2015, 18, 545-553, 10.1038/gim.2015.141.
- Marina T. DiStefano; on behalf of the ClinGen Hearing Loss Clinical Domain Working Group; Sarah E. Hemphill Ba; Andrea M. Oza; Rebecca K. Siegert Bs; Andrew R. Grant Ba; Madeline Y. Hughes Ba; Brandon J. Cushman Ba; Hela Azaiez; Kevin T. Booth Bs; et al.Alex ChapinHatice DuzkaleTatsuo Matsunaga MdJun ShenWenying ZhangMargaret KennaLisa A. SchimmentiMustafa TekinHeidi L. RehmAhmad N. Abou TayounSami S. Amr ClinGen expert clinical validity curation of 164 hearing loss gene–disease pairs. Genetics in Medicine 2019, 21, 2239-2247, 10.1038/s41436-019-0487-0.
- Ahmad N. Abou Tayoun; Bryan Krock; Nancy B. Spinner; Sequencing-based diagnostics for pediatric genetic diseases: progress and potential. Expert Review of Molecular Diagnostics 2016, 16, 987-999, 10.1080/14737159.2016.1209411.
- Sami S. Amr; Elissa Murphy; Elizabeth Duffy; Rojeen Niazi; Jorune Balciuniene; Minjie Luo; Heidi L. Rehm; Ahmad N. Abou Tayoun; Allele-Specific Droplet Digital PCR Combined with a Next-Generation Sequencing-Based Algorithm for Diagnostic Copy Number Analysis in Genes with High Homology: Proof of Concept Using Stereocilin. Clinical Chemistry 2018, 64, 705-714, 10.1373/clinchem.2017.280685.
- Stefan Rentas; Ahmad Abou Tayoun; Utility of droplet digital PCR and NGS-based CNV clinical assays in hearing loss diagnostics: current status and future prospects. Expert Review of Molecular Diagnostics 2021, 21, 213-221, 10.1080/14737159.2021.1887731.
- Ahmad N. Abou Tayoun; Christopher D. Tunkey; Trevor J. Pugh; Tristen Ross; Minita Shah; Clarence C. Lee; Timothy T. Harkins; Wendy A. Wells; Laura J. Tafe; Christopher I. Amos; et al.Gregory J. Tsongalis A Comprehensive Assay for CFTR Mutational Analysis Using Next-Generation Sequencing. Clinical Chemistry 2013, 59, 1481-1488, 10.1373/clinchem.2013.206466.
- Ahmad N. Abou Tayoun; Heather Mason-Suares; Ashley L. Frisella; Mark Bowser; Elizabeth Duffy; Lisa Mahanta; Birgit Funke; Heidi L. Rehm; Sami S. Amr; Targeted Droplet-Digital PCR as a Tool for Novel Deletion Discovery at the DFNB1 Locus. Human Mutation 2015, 37, 119-126, 10.1002/humu.22912.
- Achondroplasia . GeneReviews® . Retrieved 2021-6-8
- Cystic Fibrosis and Congenital Absence of the Vas Deferens . GeneReviews® . Retrieved 2021-6-8
- Michael S. Watson; Garry R. Cutting; Robert J. Desnick; Deborah A. Driscoll; Katherine Klinger; Michael Mennuti; Glenn E. Palomaki; Bradley W. Popovich; Victoria M. Pratt; Elizabeth M. Rohlfs; et al.Charles M. StromC. Sue RichardsDavid R. WittWayne W. Grody Cystic fibrosis population carrier screening: 2004 revision of American College of Medical Genetics mutation panel. Genetics in Medicine 2004, 6, 387-391, 10.1097/01.gim.0000139506.11694.7c.
- Keith D. Robertson; DNA methylation and human disease. Nature Reviews Genetics 2005, 6, 597-610, 10.1038/nrg1655.
- Ignatia B. Van Den Veyver; Christine M. Eng; Genome-Wide Sequencing for Prenatal Detection of Fetal Single-Gene Disorders. Cold Spring Harbor Perspectives in Medicine 2015, 5, a023077, 10.1101/cshperspect.a023077.
- David T. Miller; ACMG Secondary Findings Working Group; Kristy Lee; Wendy K. Chung; Adam S. Gordon; Gail E. Herman; Teri E. Klein; Douglas R. Stewart; Laura M. Amendola; Kathy Adelman; et al.Sherri J. BaleMichael H. GollobSteven M. HarrisonRay E. HershbergerKent McKelveyC. Sue RichardsChristopher N. VlangosMichael S. WatsonChrista Lese Martin ACMG SF v3.0 list for reporting of secondary findings in clinical exome and genome sequencing: a policy statement of the American College of Medical Genetics and Genomics (ACMG). Genetics in Medicine 2021, 24, 1-10, 10.1038/s41436-021-01172-3.
- Eric J. Duncavage; Molly C. Schroeder; Michele O’Laughlin; Roxanne Wilson; Sandra MacMillan; Andrew Bohannon; Scott Kruchowski; John Garza; Feiyu Du; Andrew E.O. Hughes; et al.Josh RobinsonEmma HughesSharon E. HeathJack D. BatyJulie NeidichMatthew J. ChristopherMeagan A. JacobyGeoffrey L. UyRobert S. FultonChristopher A. MillerJacqueline E. PaytonDaniel C. LinkMatthew J. WalterPeter WesterveltJohn F. DiPersioTimothy J. LeyDavid H. Spencer Genome Sequencing as an Alternative to Cytogenetic Analysis in Myeloid Cancers. New England Journal of Medicine 2021, 384, 924-935, 10.1056/nejmoa2024534.
- Leslie G. Biesecker; Robert C. Green; Diagnostic Clinical Genome and Exome Sequencing. New England Journal of Medicine 2014, 370, 2418-2425, 10.1056/nejmra1312543.
- G M Church; The Personal Genome Project. Molecular Systems Biology 2005, 1, 2005.0030-3, 10.1038/msb4100040.
- Emilie LaLonde; Stefan Rentas; Fumin Lin; Matthew C. Dulik; Cara M. Skraban; Nancy B. Spinner; Genomic Diagnosis for Pediatric Disorders: Revolution and Evolution. Frontiers in Pediatrics 2020, 8, 373, 10.3389/fped.2020.00373.
- Ahmad N. Abou Tayoun; Heidi L. Rehm; Genetic variation in the Middle East—an opportunity to advance the human genetics field. Genome Medicine 2020, 12, 1-4, 10.1186/s13073-020-00821-7.
- Samuel W. Baker; Jill R. Murrell; Addie I. Nesbitt; Kieran B. Pechter; Jorune Balciuniene; Xiaonan Zhao; Zhenming Yu; Elizabeth H. Denenberg; Elizabeth T. DeChene; Alisha B. Wilkens; et al.Elizabeth J. BhojQiaoning GuanMatthew C. DulikLaura K. ConlinAhmad N. Abou TayounMinjie LuoChao WuKajia CaoMahdi SarmadyEmma C. BedoukianJennifer TarpinianLivija MedneCara M. SkrabanMatthew A. DeardorffIan D. KrantzBryan L. KrockAvni B. Santani Automated Clinical Exome Reanalysis Reveals Novel Diagnoses. The Journal of Molecular Diagnostics 2019, 21, 38-48, 10.1016/j.jmoldx.2018.07.008.
- Mahdi Sarmady; Ahmad Abou Tayoun; Need for Automated Interactive Genomic Interpretation and Ongoing Reanalysis. JAMA Pediatrics 2018, 172, 1113-1114, 10.1001/jamapediatrics.2018.2675.
- Pengfei Liu; Linyan Meng; Elizabeth A. Normand; Fan Xia; Xiaofei Song; Andrew Ghazi; Jill Rosenfeld; Pilar L. Magoulas; Alicia Braxton; Patricia Ward; et al.Hongzheng DaiBo YuanWeimin BiRui XiaoXia WangTheodore ChiangFrancesco VetriniWeimin HeHanyin ChengJie DongCharul GijavanekarPaul J. BenkeJonathan A. BernsteinTanya EbleYasemen ErogluDeanna ErwinLuis EscobarJames B. GibsonKaren W. GrippSoledad KleppeMary Kay KoenigAndrea M. LewisMarvin NatowiczPedro ManciasLakeesha MinorFernando ScagliaChristian P. SchaafHaley StreffHilary VernonCrescenda L. UhlesElaine H. ZackaiNan WuV. Reid SuttonArthur L. BeaudetDonna MuznyRichard A. GibbsJennifer E. PoseySeema LalaniChad ShawChristine M. EngJames R. LupskiYaping Yang Reanalysis of Clinical Exome Sequencing Data. New England Journal of Medicine 2019, 380, 2478-2480, 10.1056/nejmc1812033.
- Branchiootorenal Spectrum Disorder. . GeneReviews® . Retrieved 2021-6-8
- Mahdi Sarmady; Ahmad Abou Tayoun; Need for Automated Interactive Genomic Interpretation and Ongoing Reanalysis. JAMA Pediatrics 2018, 172, 1113-1114, 10.1001/jamapediatrics.2018.2675.
- Pengfei Liu; Linyan Meng; Elizabeth A. Normand; Fan Xia; Xiaofei Song; Andrew Ghazi; Jill Rosenfeld; Pilar L. Magoulas; Alicia Braxton; Patricia Ward; et al.Hongzheng DaiBo YuanWeimin BiRui XiaoXia WangTheodore ChiangFrancesco VetriniWeimin HeHanyin ChengJie DongCharul GijavanekarPaul J. BenkeJonathan A. BernsteinTanya EbleYasemen ErogluDeanna ErwinLuis EscobarJames B. GibsonKaren W. GrippSoledad KleppeMary Kay KoenigAndrea M. LewisMarvin NatowiczPedro ManciasLakeesha MinorFernando ScagliaChristian P. SchaafHaley StreffHilary VernonCrescenda L. UhlesElaine H. ZackaiNan WuV. Reid SuttonArthur L. BeaudetDonna MuznyRichard A. GibbsJennifer E. PoseySeema LalaniChad ShawChristine M. EngJames R. LupskiYaping Yang Reanalysis of Clinical Exome Sequencing Data. New England Journal of Medicine 2019, 380, 2478-2480, 10.1056/nejmc1812033.
- Branchiootorenal Spectrum Disorder. . GeneReviews® . Retrieved 2021-6-8