1. Introduction
Cancer is the major cause of death worldwide. Although surgery, radiation, and chemotherapy represent the three pillars of cancer therapy, the prognosis still remains poor in advanced stages of cancer often with metastases
[1]. Thus, cancer immunotherapy, which aims to treat cancer by enhancing or inducing host immune responses to tumor cells, has long been hoped to become the fourth pillar of treatment. Historically, the potential therapeutic effect of host immune activity against cancer was first noted in the 19th century by Wilhelm Busch and Friedrich Fehleisen, who independently reported cancer regression after erysipelas infection
[2]. Subsequently, William Coley developed Coley’s Toxin, a cocktail of killed bacteria, and used it to treat cancers such as osteosarcoma and lymphoma
[2][3][2,3]. After that, Thomas and Burnet advanced the idea and proposed the cancer immunosurveillance hypothesis
[2][4][5][2,4,5]. For decades, however, it remained difficult to induce or enhance host immune responses against tumor cells. Clinical trials such as cytokine-based immunotherapies or autologous/allogenic adoptive immune cell transfers were mostly disappointing
[6]. Cancer vaccines that involve exogenous administration of tumor antigens with adjuvants to induce or enhance tumor-specific immune responses have also been tried, but mostly with unsatisfactory results
[6]. Meanwhile, the concept of cancer immunosurveillance has also evolved into the theory of cancer immunoediting, which provides three phases for the complex interactions between tumors and the host immune system; namely, elimination, equilibrium, and escape
[7][8][7,8]. Thus, it is now considered that, through these phases, tumor immunogenicity is edited and immunosuppressive mechanisms are acquired. However, the advent of immune checkpoint inhibitors has now revolutionized the field of cancer immunotherapy
[2]. Furthermore, adoptive cell therapy employing autologous T cells with synthetic chimeric antigen receptors (CARs) has also been providing highly promising clinical results
[9].
Immune checkpoint molecules negatively regulate immune responses to maintain immune homeostasis by preventing overactivated immune responses or autoimmune responses
[10][11][10,11]. In this context, cancer cells often utilize the immune checkpoint pathways to suppress host antitumor immune responses
[10][11][10,11]. Thus, blocking the immune checkpoint molecules was considered as a strategy to enhance host immune responses to tumor cells. Indeed, the development of checkpoint inhibitors, such as anti-CTLA4
[12], anti-PD-1
[13], and anti-PD-L1
[14], has brought remarkable success in cancer immunotherapy. However, it has also been demonstrated that not all cancer patients favorably respond to the immune checkpoint inhibitors, possibly due to insufficient CD8
+ cytotoxic T-lymphocytes (CTLs) against tumor cells in the host
[10][11][10,11]. Thus, additional strategies may be needed to elicit tumor antigen-specific CD8
+ CTLs in patients who do not sufficiently benefit from immune checkpoint inhibitors.
It is now known that tumor cells express endogenous tumor antigens such as tumor-associated antigens (TAAs) (such as aberrantly expressed developmentally regulated antigens) and neoantigens generated by somatic mutations
[15]. Therefore, if tumor antigen-specific CTLs are elicited in the host, they should be able to recognize and remove tumor cells. Thus, cancer vaccines are designed to induce tumor antigen-specific immune responses, particularly CD8
+ CTLs
[16]. Because dendritic cells (DCs) are professional antigen-presenting cells and have the unique ability to link innate and adaptive immunity, they have been regarded as the key target cells for cancer vaccine development. It is known that, upon antigen capture, DCs in peripheral tissues migrate to the draining lymph nodes where they present antigens to naive CD4
+ and CD8
+ T cells, resulting in the induction of effector T cells
[17][18][17,18]. In the case of intracellular antigens, DCs process them and present antigen peptides through the major histocompatibility complex (MHC) class I pathway
[17][18][19][17,18,19]. This leads to the induction of CD8
+ CTLs. In the case of extracellular antigens, DCs usually process them and present antigen peptides through the MHC class II pathway that preferentially activates CD4
+ T cells
[18][19][18,19]. To induce CD8
+ CTLs to extracellular antigens, therefore, the process called cross-presentation is needed to present antigen peptides via MHC class I
[17]. Thus, cancer vaccines need to be preferentially processed by cross-presenting DCs. To this end, a number of adjuvants have been developed to activate cross-presenting DCs and have been shown to induce strong CD8
+ CTL responses in animal models, yet very few have met the safety and efficacy requirement for human use
[18]. Thus, it is of great importance to develop new vaccines and adjuvants that efficiently promote cross-presentation of extracellular antigens including tumor antigens for specific CD8
+ CTL responses.
Recent research progress has shown that there are several DC subsets with different roles in the induction of antigen-specific immune responses
[18][20][21][18,20,21]. Among the DC subsets, conventional DC1s (cDC1s) are the ones that have the ability to cross-present extracellular antigens to naive CD8
+ T cells
[22][23][24][22,23,24]. Thus, the success of therapeutic cancer vaccines is now considered to depend on selective and efficient antigen delivery to cross-presenting cDC1s.
2. DC Subsets and Their Functions
DCs were originally recognized for their remarkable capacity to present antigens to T cells
[25][26][25,26]. Thus, DCs are regarded as the primary professional antigen-presenting cells that serve as a major link between innate and adaptive immune responses. Upon antigen capture, DCs undergo a maturation process in which they upregulate the expression of MHC molecules, costimulatory molecules (CD80/86), cytokines, chemokines, and the chemokine receptor CCR7
[27][28][27,28]. Consequently, mature DCs migrate to regional lymphoid tissues via CCR7 and activate naïve T cells to differentiate to various effector T cells, including T helper (Th) 1 cells, Th2 cells, Th17 cells, T follicular helper (T
FH) cells, regulatory T (Treg) cells, and CD8
+ CTLs, resulting in various T-cell responses
[19][27][28][19,27,28]. In this context, experimental evidence accumulated over the last two decades has revealed that DCs are heterogenous and there are different DC subsets that are specialized in priming different types of effector T cells
[20]. Thus, individual DC subsets are able to skew immune responses according to their induction of different effector cell responses. Almost all DC subsets in humans and mice are known to express CD11c and can be distinguished by differential expression of cell surface markers and their immunological functions
[18][20][21][18,20,21]. DCs are now broadly divided into four major subsets; namely, conventional DCs (cDCs), plasmacytoid DCs (pDCs), monocyte-derived DCs (moDCs), and Langerhans cells (LCs). cDCs are further subdivided into cDC1s and cDC2s. Analogous subsets have been identified in humans and mice
[18][20][21][18,20,21]. The main phenotypic and functional characteristics of these five DC subsets are described below and summarized in .
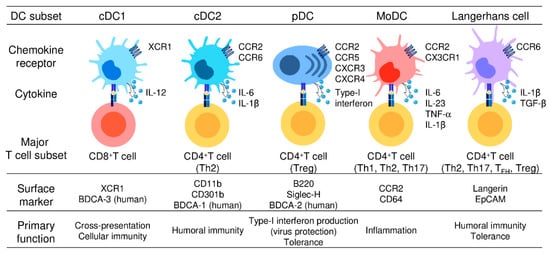
Figure 1. Different subsets of dendritic cells.
Five DC subsets are characterized by their surface phenotypes and functional properties: cDC1s (the signature markers: XCR1 and BDCA-3), cDC2s (the signature markers: CD11b, CD301b, and BDCA-1), pDCs (the signature markers: B220, Siglec-H, and BDCA-2), moDCs (the signature markers: CCR2 and CD64), and Langerhans cells (the signature markers: Langerin and EpCAM). cDC1s are specialized for CD8+ CTL and Th1 cell induction and thus mediate cellular immune responses. cDC2s present antigens to both CD4+ T cells and CD8+ T cells but preferentially induce CD4+ Th2 induction and are involved in humoral immune responses. pDCs abundantly produce type-I interferon and are critically involved in the induction of antiviral immune responses, especially in the gut. moDCs share functional characteristics with cDC1s and cDC2s, present antigens to CD4+ T cells and CD8+ T cells, and are widely involved in inflammatory responses. LCs present antigens to both CD4+ T cells and CD8+ T cells, but preferentially promote the differentiation of naïve CD4+ T cells into Th2cells, TFH cells, or Treg cells.
3. Use of DEC205 and CLEC9A in cDC1-Targeting Vaccines
Considerable efforts have been made to develop cancer vaccines that induce CD8
+ CTL responses against cancer, but no sufficiently effective cancer vaccines are available so far. Because DCs were known to play a pivotal role in the induction of various immune responses, including CD8
+ CTL responses, ex vivo-derived moDCs were also tried as DC-based cancer vaccines, but clinical outcomes were poor, possibly due to a limited capacity of in vitro-derived DCs to induce CD8
+ CTL responses.
Because recent studies have identified cDC1s as the key cross-presenting DC subset capable of inducing CD8
+ CTLs
[29][30][31][32,33,34], efforts have been focused on how to target cDC1s in antigen delivery. Because DEC-205 and CLEC9A are known to be highly expressed on cDC1s compared with other DC subsets (summarized in )
[32][33][34][35][36][37][38][39][40][41][42][43][44][45][46][47][48][49][106,107,108,109,110,111,112,113,114,115,116,117,118,119,120,121,122,123], these molecules have been used as a possible target molecule to selectively deliver antigens to cDC1s
[50][124]. DEC-205 is a C-type lectin receptor that acts as a recognition receptor for apoptotic and necrotic cells
[51][52][125,126]. Although DEC-205 is expressed on all DC subsets, it is highly expressed on cDC1s and cDC2s
[51][52][53][54][55][125,126,127,128,129]. DEC-205 is also reported to be weakly expressed on macrophages, NK cells, T cells, and B cells
[51][52][53][54][55][125,126,127,128,129]. It was shown that coadministration of antigen-conjugated anti-DEC-205 antibodies with a combination of adjuvants such as complete Freund’s adjuvant, poly (I:C), or anti-CD40 antibody, efficiently induced antigen-specific IgG responses and CD8
+ CTL responses
[32][33][34][35][106,107,108,109]. Thus, antigen-conjugated anti-DEC-205 antibodies appeared to be capable of inducing humoral immune responses by cDC2s and cytotoxic immune responses by cDC1s. However, antigen-conjugated anti-DEC-205 antibodies alone without adjuvants failed to induce CD8
+ CTL responses but rather induced immune tolerance
[34][36][108,110]. CLEC9A is another C-type lectin receptor identified as the first receptor responsible for sensing necrotic cells
[56][57][58][130,131,132]. CLEC9A is highly expressed on cDC1s and weakly expressed on pDCs and monocytes
[59][60][40,133]. CLEC9A has also been shown to promote cross-presentation of dead cell-associated antigens
[61][62][134,135]. Coadministration of antigen-conjugated anti-CLECA9 antibodies with anti-CD40 antibody efficiently induced CD8
+ CTL responses against murine melanoma
[37][38][39][40][111,112,113,114]. In the absence of adjuvants, however, antigen-conjugated anti-CLECA9 antibodies induced antigen-specific IgG responses, but not CD8
+ CTL responses, and rather strongly induced Treg cell responses
[41][42][43][115,116,117]. Thus, although DEC-205 and CLEC9A are promising surface molecules for targeted delivery of antigens to cDC1s, the vaccines based on these molecules require adjuvants for the efficient induction of CD8
+ CTL responses. Furthermore, because DEC-205 and CLEC9A are also expressed on many other immune cells, their responses may be suppressive to CD8
+ CTL responses.
Table 1. Target molecules on cDC1s for CTL-inducing adjuvants.
|
Target Molecule
|
Type
|
Expressing Cell
|
Function
|
Application (Ref)
|
|
DEC-205
|
C-type lectin receptor
|
cDC1s, cDC2s, B cells, T cells, NK cells
|
Antigen recognition (apoptotic and necrotic cells)
|
[32][33][34][35][36][106,107,108,109,110]
|
|
CLEC9A
|
C-type lectin receptor
|
cDC1s, some pDCs, monocytes
|
Antigen recognition/endocytosis (cross-presentation)
|
[37][38][39][40][41][42][43][111,112,113,114,115,116,117]
|
|
XCR1
|
Chemokine receptor
|
cDC1s
|
Cell migration
|
[44][45][46][47][48][49][118,119,120,121,122,123]
|
4. Differential Expression of Chemokine Receptors by DC Subsets
Chemokines are a large family of small structurally related chemotactic cytokines that attract various leukocytes to their source of production via corresponding receptors
[63][64][136,137]. Humans have around 50 chemokines, which are grouped into four subfamilies (CXC, CC, (X)C, and CX3C) by the motifs of the N-terminal conserved cysteine residues. Chemokine receptors belong to the seven-membrane G-protein-coupled receptor family, and there are 18 signal-transducing receptors in humans
[63][64][136,137]. Chemokines play important roles in various biological processes, such as homeostatic migration and homing of lymphocytes, inflammatory infiltration of leukocytes, cell migration and homing during development, angiogenesis, wound healing, and even cancer metastasis
[63][64][65][136,137,138].
Chemokine receptors are known to be differentially expressed on various DC subsets. For example, immature DCs express a wide variety of chemokine receptors such as CCR1, CCR2, CCR5, and CCR6, consistent with their ability to respond to a wide range of chemokines
[66][139]. Upon antigen capture, however, immature DCs downregulate the expression of these chemokine receptors and upregulate CCR7, which leads them to draining lymph nodes and other secondary lymphoid tissues where its ligands CCL19 and CCL21 are constitutively produced
[67][68][140,141]. Furthermore, cDC1s selectively express XCR1
[69][70][35,38]; cDC2s mainly express CCR2 and CCR6
[71][72][57,142]; pDC mainly express CCR2, CCR5, CXCR3, and CXCR4
[73][74][75][143,144,145]; and moDCs mainly express CCR2 and CX3CR1
[76][77][146,147]. Thus, the chemokine receptors are useful for the characterization of various DC subsets, and they may also be possible candidates for targeted delivery of antigens to specific DC subsets. Chemokines may also be used as an adjuvant to attract specific DCs at the site of vaccination.