Understanding the molecular mechanisms underlying prostate cancer (PCa) progression towards its most aggressive, castration-resistant (CRPC) stage is urgently needed to improve the therapeutic options for this almost incurable pathology. Interestingly, CRPC is known to be characterized by a peculiar hormonal landscape. It is now well established that the androgen/androgen receptor (AR) axis is still active in CRPC cells. The persistent activity of this axis in PCa progression has been shown to be related to different mechanisms, such as: intratumoral androgen synthesis, AR amplification and mutations, AR mRNA alternative splicing, increased expression/activity of AR-related transcription factors and coregulators. A deeper clarification of the expression and activities of the androgen/AR axis in the CRPC stage will likely lead to the identification of novel predictive biomarkers as well as to the improvement of the therapeutical options for this almost untreatable disease, in terms of precision medicine.
Understanding the molecular mechanisms underlying prostate cancer (PCa) progression towards its most aggressive, castration-resistant (CRPC) stage is urgently needed to improve the therapeutic options for this almost incurable pathology. Interestingly, CRPC is known to be characterized by a peculiar hormonal landscape. It is now well established that the androgen/androgen receptor (AR) axis is still active in CRPC cells. The persistent activity of this axis in PCa progression has been shown to be related to different mechanisms, such as: intratumoral androgen synthesis, AR amplification and mutations, AR mRNA alternative splicing, increased expression/activity of AR-related transcription factors and coregulators. A deeper clarification of the expression and activities of the androgen/AR axis in the CRPC stage will likely lead to the identification of novel predictive biomarkers as well as to the improvement of the therapeutical options for this almost untreatable disease, in terms of precision medicine.
- castration-resistant prostate cancer
- androgens
- androgen receptors
- AR
1. Introduction
Prostate cancer (PCa) still remains the second leading cause of cancer-related deaths in Western countries, although a higher survival rate and a long-term decline in mortality have been recently reported [1]. Most PCas are androgen-dependent in their early stage, and androgen deprivation therapy (ADT), aimed to reduce the circulating levels of testosterone and achieved by chemical castration, still represents the standard care of treatment [2,3,4][2][3][4]. Gonadotropin-releasing hormone (GnRH) analogs (agonists and antagonists), responsible for the suppression of testicular androgen production, are often associated with inhibitors of androgen receptor (AR) activity to obtain a maximal androgen deprivation condition (combined androgen blockade, CAB) [5,6][5][6]. Unfortunately, within 2–3 years, most patients progress towards the so called castration-resistant prostate cancer (CRPC) stage, characterized by tumor growth, even in the presence of castration levels of circulating androgens [7,8][7][8].
It is now well established that the androgen/androgen receptor (AR) axis remains a key player in the growth of CRPC [9,10,11,12,13][9][10][11][12][13]. Moreover, not only steroids but also peptide hormones and their receptors are deeply involved in the process of PCa progression. Gonadotropin-releasing hormone (GnRH) is the hypothalamic decapeptide known to be a key player in the functions of the pituitary gonadal axis through the activation of its pituitary receptor (GnRH-R) [14,15,16,17,18,19][14][15][16][17][18][19]. GnRH and GnRH-Rs are also expressed in different cancer cells and tissues, including PCa, both androgen-dependent and castration-resistant; the activation of these receptors is associated with a significant antitumor activity [20,21,22,23,24,25,26,27,28,29,30,31,32,33,34][20][21][22][23][24][25][26][27][28][29][30][31][32][33][34].
2. The Androgen/AR Axis in CRPC
Second-line antiandrogen therapy is indicated for the treatment of CRPC patients, supporting the idea that the androgen/AR axis is still active in this progression phase of the tumor [35,36,37,38,39,40,41][35][36][37][38][39][40][41]. To date, several mechanisms involving a persistent activity of the androgen/AR axis in CRPC have been elucidated [12,13,42,43,44][12][13][42][43][44].
2.1. Intratumoral Synthesis of Androgens
About two decades ago, it was reported that, after ADT therapy, intratumoral levels of androgens remain high in spite of serum castration levels of testosterone [45]. This observation suggested that circulating adrenal androgens might be uptaken by CRPC cells to be converted to testosterone and dihydrotestosterone (DHT). Subsequently, it was demonstrated that CRPC tissues overexpress both 3β-HSD, the enzyme responsible for the conversion of the adrenal steroid dehydroepiandrosterone (DHEA) to androstenedione, and AKR1C3 (17β-HSD), the enzyme involved in the conversion of androstenedione to testosterone and DHT and, therefore, to active androgens [46,47,48][46][47][48]. Transcription factors regulating the expression of genes involved in androgen biosynthesis are also expressed in CRPC cells [49].
It has also been suggested that, in CRPC tissues, androgens can be synthesized from cholesterol; however, this issue is still a matter of debate [48,50][48][50]. CYP17A1, the enzyme involved in the synthesis of DHEA and androstenedione, is expressed not only in the adrenal gland but also in PCa tissue [47,50][47][50]; treatment options for CRPC patients presently include abiraterone, a specific inhibitor of CYP17A1 activity [51,52,53][51][52][53].
2.2. Androgen Receptor Amplification
CRPC tissues (about 80%) from patients who progressed after ADT express high levels of AR; 30–50% of these tissues were reported to carry AR amplification, due to the presence of a high AR gene copy number [54,55,56][54][55][56]. AR amplification was also detected in circulating tumor cells (CTCs) from patients with metastatic PCa [57]. The overexpressed receptor is sensitive to low levels of androgens, thus allowing PCa cells to progress towards the CRPC stage. Interestingly, AR amplification has been found to be more common in enzalutamide than in abiraterone-resistant patients [58,59][58][59].
2.3. Androgen Receptor Mutations
The AR gene, belonging to the steroid hormone receptor superfamily, composed of eight exons, is mapped on chromosome Xp11-12 and encodes a 110 kDa (920 amino acids) protein. The full-length AR protein consists of three domains: the NH2-terminal transactivation domain (NTD, encoded by exon 1), the central and conserved DNA-binding domain (DBD, encoded by exons 2 and 3), a flexible hinge region containing a nuclear localization signal and the COOH-terminal ligand-binding domain (LBD, encoded by exons 4–8). The NTD domain contains two trinucleotide repeats that encode polyglutamine and polyglycine tracts. The interaction between NTD and LBD is necessary for the receptor transcriptional activity. In the absence of androgens, the AR is located in the cytoplasm, where it is present in an inactive conformation associated with heat shock proteins. Once bound by androgens (testosterone, DHT), the receptor translocates into the nucleus, where it is activated through dimerization, recruitments of coregulatory/epigenetic factors and stimulation of specific target genes [12,13,43,60,61,62][12][13][43][60][61][62].
Gain-of-function mutations of the AR are quite frequent (about 50%) in CRPC patients after antiandrogen therapy; these are usually single point mutations occurring mostly in the LBD of the receptor [63,64][63][64]. The T878A mutation, with alanine being substituted by threonine, was shown to confer resistance to both first- and second-generation antiandrogens (enzalutamide, apalutamide, darolutamide); similar observations were reported for the H875Y, W742C and F876L mutations [58,65,66,67,68,69][58][65][66][67][68][69]. Interestingly, by binding to these mutant receptors, antiandrogens can induce their activation, thus behaving as AR agonists; moreover, these mutations were also shown to be activated by different steroids, such as adrenal androgens, progesterone and estrogens [70,71,72][70][71][72]. Consequently, AR mutations are responsible for the continuous activation of the receptor even in the presence of low circulating androgen levels after ADT therapy, thus playing a key role in tumor progression. Some gain of function mutations were shown to favor the recruitment of coregulators to the promoter region of AR target genes, thus increasing the transcriptional activity of this receptor [73,74][73][74].
Recently, mutant AR receptor genes were reported to be easily detectable in cell-free DNA (cfDNA) obtained from CRPC patients. These data strongly support that the presence of specific circulating mutant ARs can represent a useful biomarker in terms of personalized therapy in CRPC [58,75,76,77][58][75][76][77].
The most frequent mutations of the AR in CRPC patient tissues and plasma (cfDNA) are reported in Figure 1.
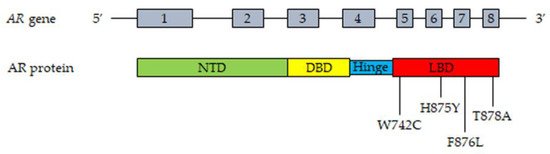
Figure 1. Most frequent mutations of AR detected in tissues and plasma cfDNA from CRPC patients. Upper part, representation of the AR gene structure. Lower part, representation of the AR protein structure with the most frequently mutated amino acids. T878A, alanine substituted by threonine; H875Y, tyrosine substituted by histidine; W742C, cysteine substituted by triptophan; F876L, leucine substituted by phenylalanine. Abbreviations: AR, androgen receptor; NTD, NH2-terminal transactivation domain; DBD, DNA-binding domain; LBD, ligand-binding domain.
2.4. Androgen Receptor Splice Variants
During the last two decades, it has become increasingly clear that, in PCa cells, the AR gene can undergo alternative splicing, giving rise to different splice variants (AR-Vs) [78,79,80,81,82][78][79][80][81][82]. Most of these variants lack the LBD but maintain the ability to enter the nucleus and to bind specific DNA response elements in a ligand-independent manner. Specifically, AR-V7 is truncated at the end of exon 3, but it has been demonstrated to be localized at the nuclear level and to retain constitutive transcriptional activity in the absence of the ligand [83]. The expression of AR-V7 was found to be associated with the development of drug resistance (enzalutamide, abiraterone) and aggressive behavior in CRPC cells [84]. In nude mice harboring human CRPC cell xenografts, treatment with abiraterone significantly increased AR-V7 expression while AR-V7 overexpression promoted tumor growth and invasiveness [85,86][85][86]; in line with this observation, targeting this receptor variant was reported to suppress tumor growth and to confer sensitivity to antiandrogens (enzalutamide) [87]. In humans, the expression of AR-V7 was observed in tumor biopsies from CRPC patients and significantly correlated with tumor progression and short survival [88,89,90,91,92][88][89][90][91][92]. High levels of AR-V7 could also be detected in extracellular vesicles purified from the plasma of CRPC patients, as well as in circulating tumor cells (CTCs) from enzalutamide- or abiraterone-resistant patients, and were found to be associated with a poor prognosis [93,94,95,96,97][93][94][95][96][97].
Additional transcript variants are generated by alternative splicing of the AR gene in CRPC [13,98,99][13][98][99]. In particular, the ARv567es splice variant originates from the loss of exons 5–7, encoding the LBD, but it conserves the hinge region of exon 4 involved in the nuclear localization of the receptor isoform, thus supporting its constitutive activity irrespective of the presence of the ligand; its expression was shown to increase in tissue biopsies after ADT therapy, and to correlate with outcomes to taxane therapy in CTCs, in PCa patients [88,100,101][88][100][101].
Mechanistically, the AR-Vs can form homodimers or heterodimers (by combining with other variants), or they can dimerize with the full-length AR. At the nuclear level, the dimers bind to response elements in the promoter region of specific downstream genes (either unique or canonical AR-regulated genes), thus modulating their expression and promoting the development of CRPC [13,42,89,91,102][13][42][89][91][102].
Based on these observations, targeting the AR-Vs and their signaling pathways might represent a novel and effective therapeutic strategy for the treatment of CRPC patients.
2.5. Androgen Receptor: Transcription Factors and Coregulators
AR-mediated gene transcription requires the interaction of the receptor with different coregulators (such as the Steroid Receptor Coactivators, SRCs) and transcription factors (such as GATA2 and FOXA1) [103,104,105,106][103][104][105][106].
The transcriptional activity of AR requires the recruitment and cooperation of transcription factors. Among these, the GATA family of transcription factors, consisting of six members, was reported to be involved in the AR-mediated signaling in CRPC cells [42,43,106][42][43][106]. In particular, GATA2 pioneer transcription factors were shown to be involved in the androgen-related regulation of PSA expression in CRPC cells; moreover, GATA protein consensus DNA sequences were observed in the AR binding regions of androgen-regulated genes in these cells, supporting their cooperation with the receptor in mediating androgen effects [106,107][106][107]. This factor was also demonstrated to be a key regulator of the transcriptional activity of AR-Vs in CRPC cells [43].
FOXA-1 (forkhead box A1) is another pioneer transcription factor involved in AR-promoted gene transcription. It was demonstrated to play a key role in AR-mediated tumor growth and progression in CRPC cells [108]. Mechanistically, FOXA1 binds to AR and the transcription factor HOXB13 at the cytoplasmic level; then, the FOXA1-AR-HOXB13 complex translocates into the nucleus where it binds, with the cooperation of GATA2, to specific DNA sequences. FOXA1, HOXB13 and GATA2 open compacted chromatin, increasing the accessibility of these DNA regions to additional transcription factors, thus promoting AR transcriptional activity and the expression of AR-regulated genes [42,108,109,110][42][108][109][110].
Coregulators modulate the activity of several proteins in the transcription complex through chemical modifications and are also involved in the recruitment of general transcription factors associated with RNA polymerase II to the constitutive promoter of target genes [106,111][106][111]. Specifically, the p160 steroid receptor coactivators (SRC-1, SRC-2 and SRC-3) promote the formation of a complex between AR enhancer sequences and the promoter region of androgen target genes, thus favoring AR transcriptional activity [112]. SRCs expression was found to positively correlate with PCa progression and recurrence [113,114,115][113][114][115]. Importantly, in PCa cells, SRC-2 was reported to interact with AR at the nuclear level to increase the sensitivity of cancer cells to androgens and to enhance the ligand-independent transcription of AR target genes [116,117][116][117]. Similarly, the AR coactivator MAGE-11 (melanoma antigen gene protein-A11) was shown to be overexpressed in CRPC cells as a consequence of the hypomethylation of CpG islands in its promoter region, providing an additional mechanism for the increased AR signaling in CRPC [118].
Taken together, these observations support the notion that the interaction of AR with specific transcription factors and coregulators plays a key role in promoting PCa growth and progression. These mechanisms are presently considered a possible molecular target for novel therapeutic approaches for CRPC.
The most relevant molecular mechanisms underlying the persistent activity of the androgen/AR axis in CRPC cells are summarized in Figure 2.
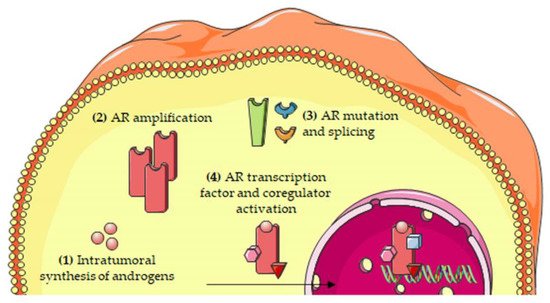
Figure 2. Molecular mechanisms involved in the persistent activity of the androgen/AR axis in CRPC cells. The persistent activation of the androgen/AR axis has been attributed to several mechanisms, including: (1) intratumoral synthesis of androgens; (2) AR amplification; (3) AR mutations and alternative splicing; and (4) increased expression/activity of transcription factors and coregulators of the AR. Abbreviations: AR, androgen receptor.
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33.
- Labrie, F.; Candas, B.; Gomez, J.L.; Cusan, L. Can combined androgen blockade provide long-term control or possible cure of localized prostate cancer? Urology 2002, 60, 115–119.
- Singer, E.A.; Golijanin, D.J.; Miyamoto, H.; Messing, E.M. Androgen deprivation therapy for prostate cancer. Expert Opin. Pharmacother. 2008, 9, 211–228.
- Litwin, M.S.; Tan, H.J. The Diagnosis and Treatment of Prostate Cancer: A Review. JAMA 2017, 317, 2532–2542.
- Onozawa, M.; Akaza, H.; Hinotsu, S.; Oya, M.; Ogawa, O.; Kitamura, T.; Suzuki, K.; Naito, S.; Namiki, M.; Nishimura, K.; et al. Combined androgen blockade achieved better oncological outcome in androgen deprivation therapy for prostate cancer: Analysis of community-based multi-institutional database across Japan using propensity score matching. Cancer Med. 2018, 7, 4893–4902.
- Tamada, S.; Iguchi, T.; Kato, M.; Asakawa, J.; Kita, K.; Yasuda, S.; Yamasaki, T.; Matsuoka, Y.; Yamaguchi, K.; Matsumura, K.; et al. Time to progression to castration-resistant prostate cancer after commencing combined androgen blockade for advanced hormone-sensitive prostate cancer. Oncotarget 2018, 9, 36966–36974.
- Perner, S.; Cronauer, M.V.; Schrader, A.J.; Klocker, H.; Culig, Z.; Baniahmad, A. Adaptive responses of androgen receptor signaling in castration-resistant prostate cancer. Oncotarget 2015, 6, 35542–35555.
- Galletti, G.; Leach, B.I.; Lam, L.; Tagawa, S.T. Mechanisms of resistance to systemic therapy in metastatic castration-resistant prostate cancer. Cancer Treat. Rev. 2017, 57, 16–27.
- Nemes, A.; Tomuleasa, C.; Kacso, G. The androgen receptor remains a key player in metastatic hormone-refractory prostate cancer. Implications for new treatments. J. BUON 2014, 19, 357–364.
- Tilki, D.; Schaeffer, E.M.; Evans, C.P. Understanding Mechanisms of Resistance in Metastatic Castration-resistant Prostate Cancer: The Role of the Androgen Receptor. Eur. Urol. Focus 2016, 2, 499–505.
- Fujimura, T.; Takayama, K.; Takahashi, S.; Inoue, S. Estrogen and Androgen Blockade for Advanced Prostate Cancer in the Era of Precision Medicine. Cancers 2018, 10, 29.
- Aurilio, G.; Cimadamore, A.; Mazzucchelli, R.; Lopez-Beltran, A.; Verri, E.; Scarpelli, M.; Massari, F.; Cheng, L.; Santoni, M.; Montironi, R. Androgen Receptor Signaling Pathway in Prostate Cancer: From Genetics to Clinical Applications. Cells 2020, 9, 2653.
- Messner, E.A.; Steele, T.M.; Tsamouri, M.M.; Hejazi, N.; Gao, A.C.; Mudryj, M.; Ghosh, P.M. The Androgen Receptor in Prostate Cancer: Effect of Structure, Ligands and Spliced Variants on Therapy. Biomedicines 2020, 8, 422.
- Baba, Y.; Matsuo, H.; Schally, A.V. Structure of the porcine LH- and FSH-releasing hormone. II. Confirmation of the proposed structure by conventional sequential analyses. Biochem. Biophys. Res. Commun. 1971, 44, 459–463.
- Schally, A.V.; Arimura, A.; Baba, Y.; Nair, R.M.; Matsuo, H.; Redding, T.W.; Debeljuk, L. Isolation and properties of the FSH and LH-releasing hormone. Biochem. Biophys. Res. Commun. 1971, 43, 393–399.
- Conn, P.M.; Crowley, W.F., Jr. Gonadotropin-releasing hormone and its analogs. Annu. Rev. Med. 1994, 45, 391–405.
- Harrison, G.S.; Wierman, M.E.; Nett, T.M.; Glode, L.M. Gonadotropin-releasing hormone and its receptor in normal and malignant cells. Endocr. Relat. Cancer 2004, 11, 725–748.
- Millar, R.P. GnRHs and GnRH receptors. Anim. Reprod. Sci. 2005, 88, 5–28.
- Tzoupis, H.; Nteli, A.; Androutsou, M.; Tselios, T. Gonadotropin Releasing Hormone and GnRH Receptor: Structure, Function and Drug Development. Curr. Med. Chem. 2019, 27, 6136.
- Limonta, P.; Marelli, M.M.; Moretti, R.M. LHRH analogues as anticancer agents: Pituitary and extrapituitary sites of action. Expert Opin. Investig. Drugs 2001, 10, 709–720.
- Limonta, P.; Moretti, R.M.; Montagnani Marelli, M.; Motta, M. The biology of gonadotropin hormone-releasing hormone: Role in the control of tumor growth and progression in humans. Front. Neuroendocrinol. 2003, 24, 279–295.
- Moretti, R.M.; Marelli, M.M.; Groeninghen, J.C.v.; Motta, M.; Limonta, P. Inhibitory activity of luteinizing hormone-releasing hormone on tumor growth and progression. Endocr. Relat. Cancer 2003, 10, 161–167.
- Marelli, M.M.; Moretti, R.M.; Caulier, J.J.; Motta, M.; Limonta, P. Gonadotropin-Releasing Hormone (GnRH) receptors in tumors: A new rationale for the therapeutical application of GnRH analogs in cancer patients? Curr. Cancer Drug Targets 2006, 6, 257–269.
- Limonta, P.; Marelli, M.M.; Mai, S.; Motta, M.; Martini, L.; Moretti, R.M. GnRH Receptors in Cancer: From Cell Biology to Novel Targeted Therapeutic Strategies. Endocr. Rev. 2012, 33, 784–811.
- Limonta, P.; Manea, M. Gonadotropin-releasing hormone receptors as molecular therapeutic targets in prostate cancer: Current options and emerging strategies. Cancer Treat. Rev. 2013, 39, 647.
- Manea, M.; Marelli, M.M.; Moretti, R.M.; Maggi, R.; Marzagalli, M.; Limonta, P. Targeting hormonal signaling pathways in castration resistant prostate cancer. Recent Pat. Anticancer Drug Discov. 2014, 9, 267–285.
- Limonta, P.; Marelli, M.M.; Moretti, R.; Marzagalli, M.; Fontana, F.; Maggi, R. GnRH in the human female reproductive axis. Vitam. Horm. 2018, 107, 27–66.
- Aguilar-Rojas, A.; Perez-Solis, M.A.; Maya-Nunez, G. The gonadotropin-releasing hormone system: Perspectives from reproduction to cancer (Review). Int. J. Oncol. 2016, 48, 861–868.
- Grundker, C.; Emons, G. The Role of Gonadotropin-Releasing Hormone in Cancer Cell Proliferation and Metastasis. Front. Endocrinol. 2017, 8, 187.
- Schally, A.V.; Block, N.L.; Rick, F.G. Discovery of LHRH and development of LHRH analogs for prostate cancer treatment. Prostate 2017, 77, 1036–1054.
- Fontana, F.; Marzagalli, M.; Montagnani Marelli, M.; Raimondi, M.; Moretti, R.M.; Limonta, P. Gonadotropin-Releasing Hormone Receptors in Prostate Cancer: Molecular Aspects and Biological Functions. Int. J. Mol. Sci. 2020, 21, 9511.
- Emons, G.; Grundker, C. The Role of Gonadotropin-Releasing Hormone (GnRH) in Endometrial Cancer. Cells 2021, 10, 292.
- Grundker, C.; Emons, G. Role of Gonadotropin-Releasing Hormone (GnRH) in Ovarian Cancer. Cells 2021, 10, 437.
- Contreras, H.R.; Lopez-Moncada, F.; Castellon, E.A. Cancer stem cell and mesenchymal cell cooperative actions in metastasis progression and hormone resistance in prostate cancer: Potential role of androgen and gonadotropinreleasing hormone receptors (Review). Int. J. Oncol. 2020, 56, 1075–1082.
- Miyake, H.; Matsushita, Y.; Watanabe, H.; Tamura, K.; Motoyama, D.; Ito, T.; Sugiyama, T.; Otsuka, A. Comparative assessment of prognostic outcomes between first-generation antiandrogens and novel androgen-receptor-axis-targeted agents in patients with non-metastatic castration-resistant prostate cancer. Int. J. Clin. Oncol. 2019, 24, 842–847.
- Altavilla, A.; Casadei, C.; Lolli, C.; Menna, C.; Ravaglia, G.; Gurioli, G.; Farolfi, A.; Brighi, N.; Conteduca, V.; Burgio, S.L.; et al. Enzalutamide for the treatment of nonmetastatic castration-resistant prostate cancer. Expert Opin. Pharmacother. 2020, 21, 2091–2099.
- Crawford, E.D.; Stanton, W.; Mandair, D. Darolutamide: An Evidenced-Based Review of Its Efficacy and Safety in the Treatment of Prostate Cancer. Cancer Manag. Res. 2020, 12, 5667–5676.
- Lavaud, P.; Dumont, C.; Thibault, C.; Albiges, L.; Baciarello, G.; Colomba, E.; Flippot, R.; Fuerea, A.; Loriot, Y.; Fizazi, K. Next-generation androgen receptor inhibitors in non-metastatic castration-resistant prostate cancer. Ther. Adv. Med. Oncol. 2020, 12, 1758835920978134.
- Mori, K.; Miura, N.; Mostafaei, H.; Quhal, F.; Sari Motlagh, R.; Pradere, B.; Kimura, S.; Kimura, T.; Egawa, S.; Briganti, A.; et al. Sequential therapy of abiraterone and enzalutamide in castration-resistant prostate cancer: A systematic review and meta-analysis. Prostate Cancer Prostatic Dis. 2020, 23, 539–548.
- Pyrgidis, N.; Vakalopoulos, I.; Sountoulides, P. Endocrine consequences of treatment with the new androgen receptor axis-targeted agents for advanced prostate cancer. Hormones 2020, 20, 73.
- Cassinello, J.; Dominguez-Lubillo, T.; Gomez-Barrera, M.; Hernando, T.; Parra, R.; Asensio, I.; Casado, M.A.; Moreno, P. Optimal treatment sequencing of abiraterone acetate plus prednisone and enzalutamide in patients with castration-resistant metastatic prostate cancer: A systematic review and meta-analysis. Cancer Treat. Rev. 2021, 93, 102152.
- Cai, Z.; Chen, W.; Zhang, J.; Li, H. Androgen receptor: What we know and what we expect in castration-resistant prostate cancer. Int. Urol. Nephrol. 2018, 50, 1753–1764.
- Feng, Q.; He, B. Androgen Receptor Signaling in the Development of Castration-Resistant Prostate Cancer. Front. Oncol. 2019, 9, 858.
- Mollica, V.; Di Nunno, V.; Cimadamore, A.; Lopez-Beltran, A.; Cheng, L.; Santoni, M.; Scarpelli, M.; Montironi, R.; Massari, F. Molecular Mechanisms Related to Hormone Inhibition Resistance in Prostate Cancer. Cells 2019, 8, 43.
- Titus, M.A.; Schell, M.J.; Lih, F.B.; Tomer, K.B.; Mohler, J.L. Testosterone and dihydrotestosterone tissue levels in recurrent prostate cancer. Clin. Cancer Res. 2005, 11, 4653–4657.
- Stanbrough, M.; Bubley, G.J.; Ross, K.; Golub, T.R.; Rubin, M.A.; Penning, T.M.; Febbo, P.G.; Balk, S.P. Increased expression of genes converting adrenal androgens to testosterone in androgen-independent prostate cancer. Cancer Res. 2006, 66, 2815–2825.
- Cai, C.; Balk, S.P. Intratumoral androgen biosynthesis in prostate cancer pathogenesis and response to therapy. Endocr. Relat. Cancer 2011, 18, R175–R182.
- Kumagai, J.; Hofland, J.; Erkens-Schulze, S.; Dits, N.F.; Steenbergen, J.; Jenster, G.; Homma, Y.; de Jong, F.H.; van Weerden, W.M. Intratumoral conversion of adrenal androgen precursors drives androgen receptor-activated cell growth in prostate cancer more potently than de novo steroidogenesis. Prostate 2013, 73, 1636.
- Montgomery, R.B.; Mostaghel, E.A.; Vessella, R.; Hess, D.L.; Kalhorn, T.F.; Higano, C.S.; True, L.D.; Nelson, P.S. Maintenance of intratumoral androgens in metastatic prostate cancer: A mechanism for castration-resistant tumor growth. Cancer Res. 2008, 68, 4447–4454.
- Mitsiades, N. A road map to comprehensive androgen receptor axis targeting for castration-resistant prostate cancer. Cancer Res. 2013, 73, 4599–4605.
- Rice, M.A.; Malhotra, S.V.; Stoyanova, T. Second-Generation Antiandrogens: From Discovery to Standard of Care in Castration Resistant Prostate Cancer. Front. Oncol. 2019, 9, 801.
- Gourdin, T. Recent progress in treating advanced prostate cancer. Curr. Opin. Oncol. 2020, 32, 210–215.
- Moussa, M.; Papatsoris, A.; Abou Chakra, M.; Sryropoulou, D.; Dellis, A. Pharmacotherapeutic strategies for castrate-resistant prostate cancer. Expert Opin. Pharmacother. 2020, 21, 1431–1448.
- Koivisto, P.; Kononen, J.; Palmberg, C.; Tammela, T.; Hyytinen, E.; Isola, J.; Trapman, J.; Cleutjens, K.; Noordzij, A.; Visakorpi, T.; et al. Androgen receptor gene amplification: A possible molecular mechanism for androgen deprivation therapy failure in prostate cancer. Cancer Res. 1997, 57, 314–319.
- Palmberg, C.; Koivisto, P.; Kakkola, L.; Tammela, T.L.; Kallioniemi, O.P.; Visakorpi, T. Androgen receptor gene amplification at primary progression predicts response to combined androgen blockade as second line therapy for advanced prostate cancer. J. Urol. 2000, 164, 1992–1995.
- Kohli, M.; Li, J.; Du, M.; Hillman, D.W.; Dehm, S.M.; Tan, W.; Carlson, R.; Campion, M.B.; Wang, L.; Wang, L.; et al. Prognostic association of plasma cell-free DNA-based androgen receptor amplification and circulating tumor cells in pre-chemotherapy metastatic castration-resistant prostate cancer patients. Prostate Cancer Prostatic Dis. 2018, 21, 411–418.
- Leversha, M.A.; Han, J.; Asgari, Z.; Danila, D.C.; Lin, O.; Gonzalez-Espinoza, R.; Anand, A.; Lilja, H.; Heller, G.; Fleisher, M.; et al. Fluorescence in situ hybridization analysis of circulating tumor cells in metastatic prostate cancer. Clin. Cancer Res. 2009, 15, 2091–2209.
- Azad, A.A.; Volik, S.V.; Wyatt, A.W.; Haegert, A.; Le Bihan, S.; Bell, R.H.; Anderson, S.A.; McConeghy, B.; Shukin, R.; Bazov, J.; et al. Androgen Receptor Gene Aberrations in Circulating Cell-Free DNA: Biomarkers of Therapeutic Resistance in Castration-Resistant Prostate Cancer. Clin. Cancer Res. 2015, 21, 2315–2324.
- Salvi, S.; Casadio, V.; Conteduca, V.; Lolli, C.; Gurioli, G.; Martignano, F.; Schepisi, G.; Testoni, S.; Scarpi, E.; Amadori, D.; et al. Circulating AR copy number and outcome to enzalutamide in docetaxel-treated metastatic castration-resistant prostate cancer. Oncotarget 2016, 7, 37839–37845.
- Gao, W.; Bohl, C.E.; Dalton, J.T. Chemistry and structural biology of androgen receptor. Chem. Rev. 2005, 105, 3352–3370.
- Matsumoto, T.; Sakari, M.; Okada, M.; Yokoyama, A.; Takahashi, S.; Kouzmenko, A.; Kato, S. The androgen receptor in health and disease. Annu. Rev. Physiol. 2013, 75, 201–224.
- Tan, M.H.; Li, J.; Xu, H.E.; Melcher, K.; Yong, E.L. Androgen receptor: Structure, role in prostate cancer and drug discovery. Acta Pharmacol. Sin. 2015, 36, 3–23.
- Taplin, M.E.; Rajeshkumar, B.; Halabi, S.; Werner, C.P.; Woda, B.A.; Picus, J.; Stadler, W.; Hayes, D.F.; Kantoff, P.W.; Vogelzang, N.J.; et al. Androgen receptor mutations in androgen-independent prostate cancer: Cancer and Leukemia Group B Study 9663. J. Clin. Oncol. 2003, 21, 2673–2678.
- Robinson, D.; Van Allen, E.M.; Wu, Y.M.; Schultz, N.; Lonigro, R.J.; Mosquera, J.M.; Montgomery, B.; Taplin, M.E.; Pritchard, C.C.; Attard, G.; et al. Integrative clinical genomics of advanced prostate cancer. Cell 2015, 161, 1215–1228.
- Taplin, M.E.; Bubley, G.J.; Shuster, T.D.; Frantz, M.E.; Spooner, A.E.; Ogata, G.K.; Keer, H.N.; Balk, S.P. Mutation of the androgen-receptor gene in metastatic androgen-independent prostate cancer. N. Engl. J. Med. 1995, 332, 1393–1398.
- Joseph, J.D.; Lu, N.; Qian, J.; Sensintaffar, J.; Shao, G.; Brigham, D.; Moon, M.; Maneval, E.C.; Chen, I.; Darimont, B.; et al. A clinically relevant androgen receptor mutation confers resistance to second-generation antiandrogens enzalutamide and ARN-509. Cancer Discov. 2013, 3, 1020–1029.
- Korpal, M.; Korn, J.M.; Gao, X.; Rakiec, D.P.; Ruddy, D.A.; Doshi, S.; Yuan, J.; Kovats, S.G.; Kim, S.; Cooke, V.G.; et al. An F876L mutation in androgen receptor confers genetic and phenotypic resistance to MDV3100 (enzalutamide). Cancer Discov. 2013, 3, 1030–1043.
- Lallous, N.; Volik, S.V.; Awrey, S.; Leblanc, E.; Tse, R.; Murillo, J.; Singh, K.; Azad, A.A.; Wyatt, A.W.; LeBihan, S.; et al. Functional analysis of androgen receptor mutations that confer anti-androgen resistance identified in circulating cell-free DNA from prostate cancer patients. Genome Biol. 2016, 17, 10.
- Prekovic, S.; van Royen, M.E.; Voet, A.R.; Geverts, B.; Houtman, R.; Melchers, D.; Zhang, K.Y.; Van den Broeck, T.; Smeets, E.; Spans, L.; et al. The Effect of F877L and T878A Mutations on Androgen Receptor Response to Enzalutamide. Mol. Cancer Ther. 2016, 15, 1702–1712.
- Miyamoto, H.; Yeh, S.; Lardy, H.; Messing, E.; Chang, C. Delta5-androstenediol is a natural hormone with androgenic activity in human prostate cancer cells. Proc. Natl. Acad. Sci. USA 1998, 95, 11083–11088.
- Zhao, X.Y.; Malloy, P.J.; Krishnan, A.V.; Swami, S.; Navone, N.M.; Peehl, D.M.; Feldman, D. Glucocorticoids can promote androgen-independent growth of prostate cancer cells through a mutated androgen receptor. Nat. Med. 2000, 6, 703–706.
- van de Wijngaart, D.J.; Molier, M.; Lusher, S.J.; Hersmus, R.; Jenster, G.; Trapman, J.; Dubbink, H.J. Systematic structure-function analysis of androgen receptor Leu701 mutants explains the properties of the prostate cancer mutant L701H. J. Biol. Chem. 2010, 285, 5097–5105.
- Steinkamp, M.P.; O’Mahony, O.A.; Brogley, M.; Rehman, H.; Lapensee, E.W.; Dhanasekaran, S.; Hofer, M.D.; Kuefer, R.; Chinnaiyan, A.; Rubin, M.A.; et al. Treatment-dependent androgen receptor mutations in prostate cancer exploit multiple mechanisms to evade therapy. Cancer Res. 2009, 69, 4434–4442.
- Barbieri, C.E.; Baca, S.C.; Lawrence, M.S.; Demichelis, F.; Blattner, M.; Theurillat, J.P.; White, T.A.; Stojanov, P.; Van Allen, E.; Stransky, N.; et al. Exome sequencing identifies recurrent SPOP, FOXA1 and MED12 mutations in prostate cancer. Nat. Genet. 2012, 44, 685–689.
- Conteduca, V.; Wetterskog, D.; Sharabiani, M.T.A.; Grande, E.; Fernandez-Perez, M.P.; Jayaram, A.; Salvi, S.; Castellano, D.; Romanel, A.; Lolli, C.; et al. Androgen receptor gene status in plasma DNA associates with worse outcome on enzalutamide or abiraterone for castration-resistant prostate cancer: A multi-institution correlative biomarker study. Ann. Oncol. 2017, 28, 1508–1516.
- Sumiyoshi, T.; Mizuno, K.; Yamasaki, T.; Miyazaki, Y.; Makino, Y.; Okasho, K.; Li, X.; Utsunomiya, N.; Goto, T.; Kobayashi, T.; et al. Clinical utility of androgen receptor gene aberrations in circulating cell-free DNA as a biomarker for treatment of castration-resistant prostate cancer. Sci. Rep. 2019, 9, 4030.
- Ledet, E.M.; Lilly, M.B.; Sonpavde, G.; Lin, E.; Nussenzveig, R.H.; Barata, P.C.; Yandell, M.; Nagy, R.J.; Kiedrowski, L.; Agarwal, N.; et al. Comprehensive Analysis of AR Alterations in Circulating Tumor DNA from Patients with Advanced Prostate Cancer. Oncologist 2020, 25, 327–333.
- Haile, S.; Sadar, M.D. Androgen receptor and its splice variants in prostate cancer. Cell Mol. Life Sci. 2011, 68, 3971–3981.
- Lu, C.; Luo, J. Decoding the androgen receptor splice variants. Translat. Androl. Urol. 2013, 2, 178–186.
- Bryce, A.H.; Antonarakis, E.S. Androgen receptor splice variant 7 in castration-resistant prostate cancer: Clinical considerations. Int. J. Urol. 2016, 23, 646–653.
- Lu, C.; Brown, L.C.; Antonarakis, E.S.; Armstrong, A.J.; Luo, J. Androgen receptor variant-driven prostate cancer II: Advances in laboratory investigations. Prostate Cancer Prostatic Dis. 2020, 23, 381–397.
- Zhu, Y.; Luo, J. Regulation of androgen receptor variants in prostate cancer. Asian J. Urol. 2020, 7, 251–257.
- Luo, J. Development of AR-V7 as a putative treatment selection marker for metastatic castration-resistant prostate cancer. Asian J. Androl. 2016, 18, 580–585.
- Li, Y.; Chan, S.C.; Brand, L.J.; Hwang, T.H.; Silverstein, K.A.; Dehm, S.M. Androgen receptor splice variants mediate enzalutamide resistance in castration-resistant prostate cancer cell lines. Cancer Res. 2013, 73, 483–489.
- Mostaghel, E.A.; Marck, B.T.; Plymate, S.R.; Vessella, R.L.; Balk, S.; Matsumoto, A.M.; Nelson, P.S.; Montgomery, R.B. Resistance to CYP17A1 inhibition with abiraterone in castration-resistant prostate cancer: Induction of steroidogenesis and androgen receptor splice variants. Clin. Cancer Res. 2011, 17, 5913–5925.
- Sarwar, M.; Semenas, J.; Miftakhova, R.; Simoulis, A.; Robinson, B.; Gjorloff Wingren, A.; Mongan, N.P.; Heery, D.M.; Johnsson, H.; Abrahamsson, P.A.; et al. Targeted suppression of AR-V7 using PIP5K1alpha inhibitor overcomes enzalutamide resistance in prostate cancer cells. Oncotarget 2016, 7, 63065–63081.
- Cao, Q.; Song, Z.; Ruan, H.; Wang, C.; Yang, X.; Bao, L.; Wang, K.; Cheng, G.; Xu, T.; Xiao, W.; et al. Targeting the KIF4A/AR Axis to Reverse Endocrine Therapy Resistance in Castration-resistant Prostate Cancer. Clin. Cancer Res. 2020, 26, 1516–1528.
- Sun, S.; Sprenger, C.C.; Vessella, R.L.; Haugk, K.; Soriano, K.; Mostaghel, E.A.; Page, S.T.; Coleman, I.M.; Nguyen, H.M.; Sun, H.; et al. Castration resistance in human prostate cancer is conferred by a frequently occurring androgen receptor splice variant. J. Clin. Investig. 2010, 120, 2715–2730.
- Hornberg, E.; Ylitalo, E.B.; Crnalic, S.; Antti, H.; Stattin, P.; Widmark, A.; Bergh, A.; Wikstrom, P. Expression of androgen receptor splice variants in prostate cancer bone metastases is associated with castration-resistance and short survival. PLoS ONE 2011, 6, e19059.
- Zhang, X.; Morrissey, C.; Sun, S.; Ketchandji, M.; Nelson, P.S.; True, L.D.; Vakar-Lopez, F.; Vessella, R.L.; Plymate, S.R. Androgen receptor variants occur frequently in castration resistant prostate cancer metastases. PLoS ONE 2011, 6, e27970.
- Welti, J.; Rodrigues, D.N.; Sharp, A.; Sun, S.; Lorente, D.; Riisnaes, R.; Figueiredo, I.; Zafeiriou, Z.; Rescigno, P.; de Bono, J.S.; et al. Analytical Validation and Clinical Qualification of a New Immunohistochemical Assay for Androgen Receptor Splice Variant-7 Protein Expression in Metastatic Castration-resistant Prostate Cancer. Eur. Urol. 2016, 70, 599–608.
- Sharp, A.; Coleman, I.; Yuan, W.; Sprenger, C.; Dolling, D.; Rodrigues, D.N.; Russo, J.W.; Figueiredo, I.; Bertan, C.; Seed, G.; et al. Androgen receptor splice variant-7 expression emerges with castration resistance in prostate cancer. J. Clin. Investig. 2019, 129, 192–208.
- Antonarakis, E.S.; Lu, C.; Wang, H.; Luber, B.; Nakazawa, M.; Roeser, J.C.; Chen, Y.; Mohammad, T.A.; Chen, Y.; Fedor, H.L.; et al. AR-V7 and resistance to enzalutamide and abiraterone in prostate cancer. N. Engl. J. Med. 2014, 371, 1028–1038.
- Strati, A.; Zavridou, M.; Bournakis, E.; Mastoraki, S.; Lianidou, E. Expression pattern of androgen receptors, AR-V7 and AR-567es, in circulating tumor cells and paired plasma-derived extracellular vesicles in metastatic castration resistant prostate cancer. Analyst 2019, 144, 6671–6680.
- Foroni, C.; Zarovni, N.; Bianciardi, L.; Bernardi, S.; Triggiani, L.; Zocco, D.; Venturella, M.; Chiesi, A.; Valcamonico, F.; Berruti, A. When Less Is More: Specific Capture and Analysis of Tumor Exosomes in Plasma Increases the Sensitivity of Liquid Biopsy for Comprehensive Detection of Multiple Androgen Receptor Phenotypes in Advanced Prostate Cancer Patients. Biomedicines 2020, 8, 131.
- Zhang, T.; Karsh, L.I.; Nissenblatt, M.J.; Canfield, S.E. Androgen Receptor Splice Variant, AR-V7, as a Biomarker of Resistance to Androgen Axis-Targeted Therapies in Advanced Prostate Cancer. Clin. Genitourin. Cancer 2020, 18, 1–10.
- Zavridou, M.; Strati, A.; Bournakis, E.; Smilkou, S.; Tserpeli, V.; Lianidou, E. Prognostic Significance of Gene Expression and DNA Methylation Markers in Circulating Tumor Cells and Paired Plasma Derived Exosomes in Metastatic Castration Resistant Prostate Cancer. Cancers 2021, 13, 780.
- Ware, K.E.; Garcia-Blanco, M.A.; Armstrong, A.J.; Dehm, S.M. Biologic and clinical significance of androgen receptor variants in castration resistant prostate cancer. Endocr. Relat. Cancer 2014, 21, T87–T103.
- Kallio, H.M.L.; Hieta, R.; Latonen, L.; Brofeldt, A.; Annala, M.; Kivinummi, K.; Tammela, T.L.; Nykter, M.; Isaacs, W.B.; Lilja, H.G.; et al. Constitutively active androgen receptor splice variants AR-V3, AR-V7 and AR-V9 are co-expressed in castration-resistant prostate cancer metastases. Br. J. Cancer 2018, 119, 347–356.
- Tagawa, S.T.; Antonarakis, E.S.; Gjyrezi, A.; Galletti, G.; Kim, S.; Worroll, D.; Stewart, J.; Zaher, A.; Szatrowski, T.P.; Ballman, K.V.; et al. Expression of AR-V7 and ARv(567es) in Circulating Tumor Cells Correlates with Outcomes to Taxane Therapy in Men with Metastatic Prostate Cancer Treated in TAXYNERGY. Clin. Cancer Res. 2019, 25, 1880–1888.
- Nagandla, H.; Robertson, M.J.; Putluri, V.; Putluri, N.; Coarfa, C.; Weigel, N.L. Isoform-specific Activities of Androgen Receptor and its Splice Variants in Prostate Cancer Cells. Endocrinology 2021, 162, bqaa227.
- Yang, J.C.; Ok, J.H.; Busby, J.E.; Borowsky, A.D.; Kung, H.J.; Evans, C.P. Aberrant activation of androgen receptor in a new neuropeptide-autocrine model of androgen-insensitive prostate cancer. Cancer Res. 2009, 69, 151–160.
- Heemers, H.V.; Tindall, D.J. Androgen receptor (AR) coregulators: A diversity of functions converging on and regulating the AR transcriptional complex. Endocr. Rev. 2007, 28, 778–808.
- Chandrasekar, T.; Yang, J.C.; Gao, A.C.; Evans, C.P. Mechanisms of resistance in castration-resistant prostate cancer (CRPC). Translat. Androl. Urol. 2015, 4, 365–380.
- Foley, C.; Mitsiades, N. Moving Beyond the Androgen Receptor (AR): Targeting AR-Interacting Proteins to Treat Prostate Cancer. Horm. Cancer 2016, 7, 84–103.
- Obinata, D.; Takayama, K.; Takahashi, S.; Inoue, S. Crosstalk of the Androgen Receptor with Transcriptional Collaborators: Potential Therapeutic Targets for Castration-Resistant Prostate Cancer. Cancers 2017, 9, 22.
- Bishop, J.L.; Thaper, D.; Vahid, S.; Davies, A.; Ketola, K.; Kuruma, H.; Jama, R.; Nip, K.M.; Angeles, A.; Johnson, F.; et al. The Master Neural Transcription Factor BRN2 Is an Androgen Receptor-Suppressed Driver of Neuroendocrine Differentiation in Prostate Cancer. Cancer Discov. 2017, 7, 54–71.
- Teng, M.; Zhou, S.; Cai, C.; Lupien, M.; He, H.H. Pioneer of prostate cancer: Past, present and the future of FOXA1. Protein Cell 2021, 12, 29–38.
- Copeland, B.T.; Pal, S.K.; Bolton, E.C.; Jones, J.O. The androgen receptor malignancy shift in prostate cancer. Prostate 2018, 78, 521–531.
- Hankey, W.; Chen, Z.; Wang, Q. Shaping Chromatin States in Prostate Cancer by Pioneer Transcription Factors. Cancer Res. 2020, 80, 2427–2436.
- Hermanson, O.; Glass, C.K.; Rosenfeld, M.G. Nuclear receptor coregulators: Multiple modes of modification. Trends Endocrinol. Metab. 2002, 13, 55–60.
- Wang, Q.; Carroll, J.S.; Brown, M. Spatial and temporal recruitment of androgen receptor and its coactivators involves chromosomal looping and polymerase tracking. Mol. Cell 2005, 19, 631–642.
- Chung, A.C.; Zhou, S.; Liao, L.; Tien, J.C.; Greenberg, N.M.; Xu, J. Genetic ablation of the amplified-in-breast cancer 1 inhibits spontaneous prostate cancer progression in mice. Cancer Res. 2007, 67, 5965–5975.
- Xu, J.; Wu, R.C.; O’Malley, B.W. Normal and cancer-related functions of the p160 steroid receptor co-activator (SRC) family. Nat. Rev. Cancer 2009, 9, 615–630.
- Taylor, B.S.; Schultz, N.; Hieronymus, H.; Gopalan, A.; Xiao, Y.; Carver, B.S.; Arora, V.K.; Kaushik, P.; Cerami, E.; Reva, B.; et al. Integrative genomic profiling of human prostate cancer. Cancer Cell 2010, 18, 11–22.
- Ueda, T.; Mawji, N.R.; Bruchovsky, N.; Sadar, M.D. Ligand-independent activation of the androgen receptor by interleukin-6 and the role of steroid receptor coactivator-1 in prostate cancer cells. J. Biol. Chem. 2002, 277, 38087–38094.
- Fujimoto, N.; Miyamoto, H.; Mizokami, A.; Harada, S.; Nomura, M.; Ueta, Y.; Sasaguri, T.; Matsumoto, T. Prostate cancer cells increase androgen sensitivity by increase in nuclear androgen receptor and androgen receptor coactivators; a possible mechanism of hormone-resistance of prostate cancer cells. Cancer Investig. 2007, 25, 32–37.
- Karpf, A.R.; Bai, S.; James, S.R.; Mohler, J.L.; Wilson, E.M. Increased expression of androgen receptor coregulator MAGE-11 in prostate cancer by DNA hypomethylation and cyclic AMP. Mol. Cancer Res. 2009, 7, 523–535.