Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Catherine Yang and Version 1 by Angela Ostuni.
Pseudoxanthoma elasticum (PXE) is a complex autosomal recessive disease caused by mutations of ABCC6 transporter and characterized by ectopic mineralization of soft connective tissues.
- ABCC6
- Pseudoxanthoma elasticum
- cancer
1. Introduction
Pseudoxanthoma elasticum (PXE) is an autosomal recessive disease, which was described in 1881 by a French dermatologist. In 2000, it was first recognized that mutation in ABCC6 is responsible for PXE [1]. It is affecting approximately 1:50,000 people worldwide, with the prominent characteristic feature of ectopic mineralization of soft tissues like skin, eyes, and arteries (Figure 1), for which no effective curative treatment is available [2,3][2][3]. Moreover, PXE shows similar phenotypic characteristics with other common health problems like kidney diseases (chronic kidney disease (CKD) and nephrocalcinosis) and cardiovascular diseases (coronary heart disease, cardiomyopathy, and dyslipidemia), which makes PXE a complex disorder [4,5][4][5].
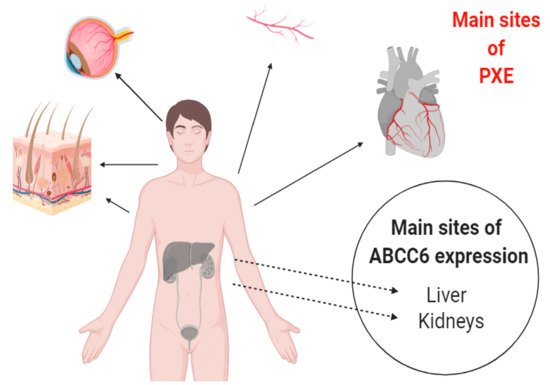
Figure 1. Pictorial presentation of ABCC6 expression and affected tissues in Pseudoxanthoma elasticum (PXE). The protein is expressed mainly in liver and kidney cells but the main areas involved in ectopic calcification are the elastic tissues of heart, blood vessels, skin, and eyes.
Different hypotheses were proposed for the factors pathologically involved with PXE. The “Metabolic Hypothesis” stated that decrease or loss of ABCC6 functionality especially in the liver may lead to a decrease in some circulating factors in the blood stream, which should be responsible for preventing ectopic mineralization of soft tissues. The “PXE Cell Hypothesis” stated that absence of ABCC6 in PXE tissues leads to an alteration in cell proliferation due to changes in the biosynthetic pathway and alters cells to extracellular matrix interactions. The most recent “ATP Release Hypothesis” stated that ABCC6 mediates the efflux of ATP in extracellular milieu, where it is hydrolyzed into AMP and pyrophosphate and prevents the mineralization of soft tissues [1].
2. Structural Properties of ABCC6 Transporter
The ATP-binding cassette (ABC) transporters are a well-known family and ubiquitously found in all living organisms. About 50 different types of ABC transporters have been recognized in humans, divided into seven subfamilies (ABCA to ABCG) based on their structure and genetic sequences. Among them we can find both half transporters, with only one transmembrane domain (TMD) and nucleotide-binding domain (NBD), and full transporters, with two TMDs and NBDs [9,10][6][7]. ABCC6 belongs to the ATP-binding cassette (ABC) transporter subfamily C and has been found to be highly expressed in basolateral plasma membrane of hepatocytes and, to some lesser extent, in the proximal tubules of the kidney [11][8].
The ABCC6 gene has 31 exons and encodes a protein of 1503 amino acid residues, MRP6. It is made-up of two nucleotide-binding domains (NBD1 and NBD2) and two transmembrane domains (TMD1 and TMD2) with an additional auxiliary NH2-terminal transmembrane domain known as TMD0, which is connected with the canonical components through the cytoplasmic L0 loop [12,13][9][10]. The NBDs of ABC transporters consist of different conserved motifs which bind and hydrolyze ATP. Like in other transporters, ABCC6 NBDs domains have Walker A motif (P loop), Walker B motif (Mg2+ binding site), histidine loop (Switch region), signature motif (C loop), Q loop (between Walker A and C-loop), and D loop (involved in the intermolecular interactions at the NBD dimmer interface). In the general process of dimerization, the Walker A/B motifs of one NBD are aligned with the C-loop of the other in a yin-yang fashion, forming a sandwich with two molecules of ATP in the middle, which are hydrolyzed in the catalytic cycle. Dimerization induces conformational changes in the TMDs, which are reversed by hydrolysis of ATP, leading to efflux of molecules [1,14,15][1][11][12]. In this situation, the ATP should be sandwiched between two sequence/structural motifs that are both rigorously conserved and form a stable homodimer [16][13]; this is not always possible with the human transporters, because one of the two NBDs is degenerate and the sequence of motifs which are responsible for hydrolysis of ATP is not identical or canonical [14][11].
In this context to elucidate the role of NBDs, different experiments were conducted on NBD1 of ABCC1/MRP1, which not only has a high level of homology with ABCC6/MRP6 but also shows a superposition of substrates [17][14]. Studies revealed that the NBDs of MRP1 are not functionally identical, as both the NBDs bind ATP but only NBD2 is involved in hydrolysis. Moreover, the ATP binding affinity of NBD1 of MRP1 is not completely dependent on NBD2 but the binding of ATP to NBD2 is highly dependent on NBD1 [18,19][15][16]. In respect of this, we have gone through by a series of experiments to identify the functional roles of NBDs, TMD0, and L0 loop of ABCC6 (Figure 2).
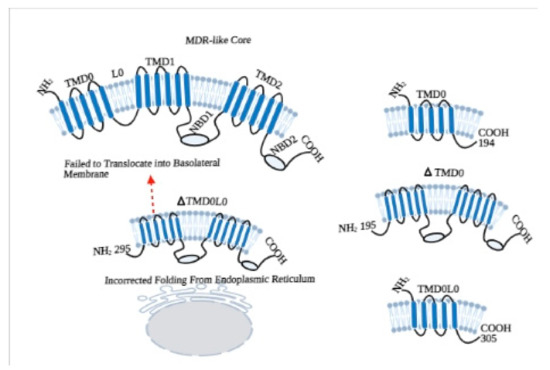
In order to evaluate the functioning of NBDs, we have first characterized the NBD1 of ABCC6/MRP6. In this experiment, by using the E748-A785 fragment of MRP6-NBD1, we have compared the helical structure and ATP binding properties of wild type and the R765Q mutated sequence, which is present in PXE patients. The study with circular dichroism analysis revealed that, in both the wild type and mutated (R765Q) NBD1, E748-A785 peptides adopted α-helical conformations, and the helical content was almost the same for both the peptides in aqueous solutions of trifluoroethanol (TFE). No differences in the length of helices have been found between the peptides in NMR spectroscopy. Moreover, Fluorescence Spectroscopy showed no significant difference in ATP binding capacity of both the peptides. These findings suggest that occurrence of PXE symptoms in R765Q mutated patients might be due to different kind of interactions [21][18].
In subsequent investigations, we have undergone two different experiments with NBD1 and NBD2, because mutational studies of PXE patients showed that domain-domain interaction is important for proper working of ABCC6 transporter and there is a functional difference between both NBDs of MRP6 [22][19]. In the first study, we constructed the full-length NBD1 (residues from Asp-627 to Leu-851) and short-length NBD1 (residues from Arg-648 to Thr-805) without some key residues; then differences in helical structure, ATP binding, and hydrolysis of both polypeptides were analyzed. Interestingly, both the polypeptides assumed predominantly α-helical conformation. However, only long-length NBD1 showed β-strand conformation, while short-length NBD1 showed higher helix content, which suggested that the sequences D627-H647 and T806-L851, which are only present in the long-length polypeptide, assume a β-strand conformation, similarly to the corresponding regions of MRP1. Although fluorescence quenching experiments revealed that both the polypeptides have the affinity to bind nucleotides (ATP and ADP), the long-length NBD1 showed a higher affinity to bind ATP than the short-length NBD. In addition, no hydrolytic activity was found in short-length NBD1 compared to long-length NBD1 during spectrophotometric analysis of ATPase activity. These findings suggest that short-length NBD1 lacks of some essential residues, which are responsible for ATP hydrolysis. Whereas, long-length NBD1 form homodimer in the presence of ATP, which is the property of half-transporters and physiologically, it is not possible with the full-transporter where NBD1 interacts with NBD2 [12][9].
In order to explore how the ABCC6 transporter works in-vivo, we have constructed long-length (Thr-1252 to Val-1503) and short-length NBD2 (Val-1295 to Arg-1468), and we created homology models for homodimer of NBD1 and NBD2, and heterodimer of (NBD1-NBD2) [23][20]. Circular dichroism spectra of long-length NBD2 revealed that it is more structured and contains α-helical conformation. The nucleotide (ATP and ADP) binding affinity of NBD1 and NBD2 were found to be the same during fluorescence quenching analysis. The ATPase activity of both homo and heterodimer were different, as the amount of inorganic phosphate (Pi) produced by NBD2 was lesser then NBD1. Moreover, addition of NBD2 reduced the NBD1 hydrolytic activity. These findings suggest that NBD2 is well structured (it contains α-helix and β-strands) and binds the nucleotides efficiently, but the ATPase activity of NBD2 is lower compared to the NBD1; this reflects that NBD2 is not able to form a functionally active homodimer, as supported by in-silico structural analysis. In addition, decreased ATPase activity of combined NBD1-NBD2 compared to NBD1 alone suggests that NBD1 and NBD2 work together to form a stable heterodimer and function in a regulated manner, whereas NBD1 alone works in an uncontrolled manner. This can be justified by the in-vivo functioning of the transporter [24][21], where ATP is hydrolyzed after the binding of substrate on TMDs and leads to conformational changes in membrane domains that activate the NBDs.
3. The Decrease or Loss of ABCC6 Functionality Changes Extracellular Environment
In early pathological investigations of PXE, Iliás et al. in 2002 proposed that ABCC6 is a transporter of organic anions and actively transports glutathione conjugates, including leukotriene C4 and N-ethylmaleimide S-glutathione (NEM-GS), and their abolishment due to missense mutation in ABCC6 gene is responsible for PXE [26,27][22][23]. In the same line of thinking, Borst and co-workers in 2008 proposed that ABCC6 transporter mediates the efflux of vitamin-K as a glutathione or glucuronide conjugate and is involved to prevent calcification of soft tissues [28][24]. However, in the further investigations, it was found that ABCC6 is not a transporter of vitamin-K, as its administration in PXE mouse model (ABCC6−/−) was not able to prevent the mineralization [29,30][25][26]. As discussed above, Jansen et al. in 2013 proposed that ABCC6 transporter mediates the efflux of ATP and indirectly produces PPi to prevent ectopic mineralization [2]. However, we have demonstrated that PXE is a complex metabolic disease with the reprogramming of crucial genetic factors in the absence of ABCC6 transporter activity [31,32,33][27][28][29].
In order to better understand the pathomechanism of PXE (Figure 43), we stably knocked down the ABCC6 gene in HepG2 cells by using shRNA, and its associated transcriptional/genetic changes were studied. We first examined the production of reactive oxygen species (ROS), which are supposed to increase according to the previous PXE fibroblast studies [34][30]. On the contrary, in the ABCC6 knockdown HepG2 cells, the ratio of GSH/GSSG has been found to be increased whereas a significant decrease in ROS level was observed, which means that knockdown cells resembled the reductive stress, which is also required by proliferating cells.
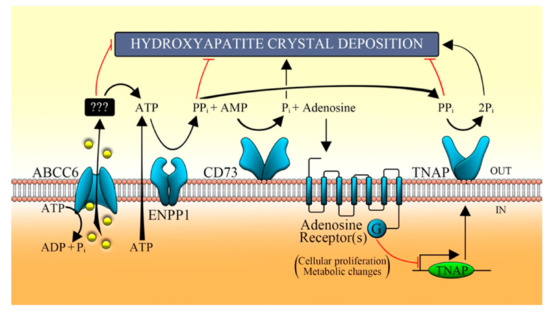
Figure 43. Proposed pathomechanism of PXE. ABCC6 transporter mediates the efflux of ATP, which is metabolized by some ecto-nucleotidases (such as ENPP1) in AMP, which in turn is converted in adenosine and Pi by CD73. In the extracellular milieu, nucleotides regulate the activity of TNAP through the purinergic pathway and prevent the ectopic mineralization [32][28]. ABCC6, ATP-binding cassette, sub-family C, member 6; ENPP1, ecto-nucleotide pyrophosphatase/phosphodiesterase type I; CD73, cluster of differentiation 73; TNAP, tissue non-specific alkaline phosphatase; Pi, inorganic phosphate; PPi, inorganic pyrophosphate [35][31].
However, we found significant delay in G1 to S transition and slower cell growth in ABCC6 knockdown HepG2 cells (Figure 54). In addition, expression of cyclin-dependent kinase inhibitor (CDKI) p21, which negatively regulates the activity of CDK and is required for the cell entry into the different cycle phases, was found increased in knockdown cells. Moreover, the expression of lamin A/C, which is required to maintain the strength of the nucleus and is pathologically involved in aging process, was decreased in those cells [33][29].
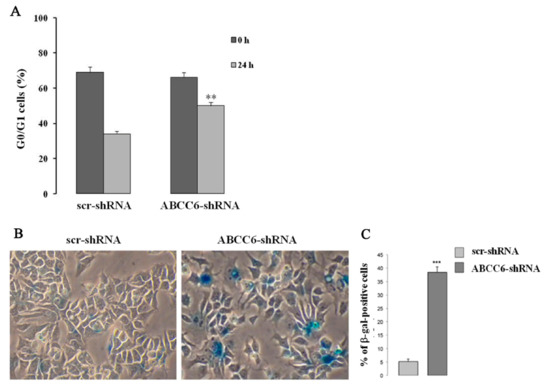
Figure 54. Pictorial presentation of senescent-like phenotype in ABCC6 knockdown HepG2 cell. (A)—For the cell cycle analysis, the cells were synchronized at the G1 phase by serum deprivation for 24 h, restimulated with serum for 24 h, and analyzed using flow cytometry after BrDU and PI staining. The percentage of control (scr-shRNA) and ABCC6 knockdown cells (ABCC6-shRNA) in G0/G1 was recorded. (B)—Representative images (40 × magnification) of senescence-associated β-galactosidase staining in control and ABCC6 knockdown cells. (C)—Quantitative analysis of positive β-galactosidase-stained cells. Data were generated from three independent experiments performed in triplicate and are shown as means ± SD. Statistical analysis was performed using unpaired Student’s t test: ** p < 0.01 and *** p < 0.001 [33][29].
Interestingly, knockdown HepG2 cells were shown to have a decreased expression of the ecto-5′-nucleotidase (NT5E or CD73), which regulates the conversion of AMP to adenosine and Pi (inorganic phosphate). Additionally, nonspecific alkaline phosphatase (TNAP), whose activity normally maintains Pi/iPP ratio accelerating the mineralization, was found to be increased in knockdown HepG2 cells. These results suggest that the absence of transporter activity in the hepatic cells decreases the NT5E expression and increases the pro-mineralizing TNAP activity, which is also clinically found in patients with arterial calcifications due to deficiency in CD73 (ACDC) [31][27]. It is clear from the knockdown study that the absence of transporter activity leads to alteration in gene expression, which is required to provide PPi to prevent mineralization. However, in a further study, we also found that ABCC6 plays a crucial role to activate the purinergic pathway, which is important to maintain proper cellular function. In this study, pharmacological inhibition of ABCC6 by probenecid down-regulated the expression of CD73. On the contrary, the expression of CD73 was found increased after the application of adenosine and ATP, which strengthens the idea that ABCC6 not only mediates the efflux of ATP but also regulates the purinergic system [35][31].
References
- Moitra, K.; Garcia, S.; Jaldin, M.; Etoundi, C.; Cooper, D.; Roland, A.; Dixon, P.; Reyes, S.; Turan, S.; Terry, S.; et al. ABCC6 and pseudoxanthoma elasticum: The face of a rare disease from genetics to advocacy. Int. J. Mol. Sci. 2017, 18, 1488.
- Jansen, R.S.; Küçükosmanoglu, A.; De Haas, M.; Sapthu, S.; Otero, J.A.; Hegman, I.E.M.; Bergen, A.A.B.; Gorgels, T.G.M.F.; Borst, P.; Van De Wetering, K. ABCC6 prevents ectopic mineralization seen in pseudoxanthoma elasticum by inducing cellular nucleotide release. Proc. Natl. Acad. Sci. USA 2013, 110, 20206–20211.
- Luo, H.; Li, Q.; Cao, Y.; Uitto, J. Therapeutics Development for Pseudoxanthoma Elasticum and Related Ectopic Mineralization Disorders: Update 2020. J. Clin. Med. 2021, 10, 114.
- De Vilder, E.Y.; Hosen, M.J.; Vanakker, O.M. The ABCC6 transporter as a paradigm for networking from an orphan disease to complex disorders. Biomed. Res. Int. 2015, 2015, 1–18.
- Lau, W.L.; Liu, S.; Vaziri, N.D. Chronic kidney disease results in deficiency of ABCC6, the novel inhibitor of vascular calcification. Am. J. Nephrol. 2014, 40, 51–55.
- Xiong, J.; Feng, J.; Yuan, D.; Zhou, J.; Miao, W. Tracing the structural evolution of eukaryotic ATP binding cassette transporter superfamily. Sci. Rep. 2015, 5, 16724.
- Liu, X.; Pan, G. Drug Transporters in Drug Disposition, Effects and Toxicity; Springer: Berlin/Heidelberg, Germany, 2019.
- Favre, G.; Laurain, A.; Aranyi, T.; Szeri, F.; Fulop, K.; Le Saux, O.; Duranton, C.; Kauffenstein, G.; Martin, L.; Lefthériotis, G. The ABCC6 transporter: A new player in biomineralization. Int. J. Mol. Sci. 2017, 18, 1941.
- Ostuni, A.; Miglionico, R.; Castiglione Morelli, M.A.; Bisaccia, F. Study of the nucleotide-binding domain 1 of the human transporter protein MRP6. Protein Pept. Lett. 2010, 17, 1553–1558.
- Ostuni, A.; Castiglione Morelli, M.A.; Cuviello, F.; Bavoso, A.; Bisaccia, F. Structural characterization of the L0 cytoplasmic loop of human multidrug resistance protein 6 (MRP6). Biochim. Biophys. Acta Biomembr. 2019, 1861, 380–386.
- Thomas, C.; Tampé, R. Multifaceted structures and mechanisms of ABC transport systems in health and disease. Curr. Opin. Struct. Biol. 2018, 51, 116–128.
- Ran, Y.; Thibodeau, P.H. Stabilization of nucleotide binding domain dimers rescues ABCC6 mutants associated with pseudoxanthoma elasticum. J. Biol. Chem. 2017, 292, 1559–1572.
- Smith, P.C.; Karpowich, N.; Millen, L.; Moody, J.E.; Rosen, J.; Thomas, P.J.; Hunt, J.F. ATP binding to the motor domain from an ABC transporter drives formation of a nucleotide sandwich dimer. Mol. Cell 2002, 10, 139–149.
- Ran, Y.; Zheng, A.; Thibodeau, P.H. Structural analysis reveals pathomechanisms associated with pseudoxanthoma elasticum–causing mutations in the ABCC6 transporter. J. Biol. Chem. 2018, 293, 15855–15866.
- Dhasmana, D.; Singh, A.; Shukla, R.; Tripathi, T.; Garg, N. Targeting nucleotide binding domain of multidrug resistance-associated protein-1 (MRP1) for the reversal of multi drug resistance in cancer. Sci. Rep. 2018, 8, 1–11.
- Johnson, Z.L.; Chen, J. Structural basis of substrate recognition by the multidrug resistance protein MRP1. Cell 2017, 168, 1075–1085.
- Miglionico, R.; Gerbino, A.; Ostuni, A.; Armentano, M.F.; Monné, M.; Carmosino, M.; Bisaccia, F. New insights into the roles of the N-terminal region of the ABCC6 transporter. J. Bioenerg. Biomembr. 2016, 48, 259–267.
- Ostuni, A.; Miglionico, R.; Bisaccia, F.; Castiglione Morelli, M.A. Biochemical characterization and NMR study of the region E748-A785 of the human protein MRP6/ABCC6. Protein Pept. Lett. 2010, 17, 861–866.
- Le Saux, O.; Beck, K.; Sachsinger, C.; Silvestri, C.; Treiber, C.; Göring, H.H.H.; Johnson, E.W.; De Paepe, A.; Pope, F.M.; Pasquali-Ronchetti, I.; et al. A spectrum of ABCC6 mutations is responsible for pseudoxanthoma elasticum. Am. J. Hum. Genet. 2001, 69, 749–764.
- Ostuni, A.; Miglionico, R.; Monné, M.; Castiglione Morelli, M.A.; Bisaccia, F. The nucleotide-binding domain 2 of the human transporter protein MRP6. J. Bioenerg. Biomembr. 2011, 43, 465.
- Goda, K.; Dönmez-Cakil, Y.; Tarapcsák, S.; Szalóki, G.; Szöllősi, D.; Parveen, Z.; Türk, D.; Szakács, G.; Chiba, P.; Stockner, T. Human ABCB1 with an ABCB11-like degenerate nucleotide binding site maintains transport activity by avoiding nucleotide occlusion. PLoS Genet. 2020, 16, e1009016.
- Iliás, A.; Urbán, Z.; Seidl, T.L.; Le Saux, O.; Sinkó, E.; Boyd, C.D.; Sarkadi, B.; Váradi, A. Loss of ATP-dependent transport activity in pseudoxanthoma elasticum-associated mutants of human ABCC6 (MRP6). J. Biol. Chem. 2002, 277, 16860–16867.
- Belinsky, M.G.; Chen, Z.-S.; Shchaveleva, I.; Zeng, H.; Kruh, G.D. Characterization of the drug resistance and transport properties of multidrug resistance protein 6 (MRP6, ABCC6). Cancer Res. 2002, 62, 6172–6177.
- Borst, P.; van de Wetering, K.; Schlingemann, R. Does the absence of ABCC6 (multidrug resistance protein 6) in patients with Pseudoxanthoma elasticum prevent the liver from providing sufficient vitamin K to the periphery? Cell Cycle 2008, 7, 1575–1579.
- Gorgels, T.G.M.F.; Waarsing, J.H.; Herfs, M.; Versteeg, D.; Schoensiegel, F.; Sato, T.; Schlingemann, R.O.; Ivandic, B.; Vermeer, C.; Schurgers, L.J.; et al. Vitamin K supplementation increases vitamin K tissue levels but fails to counteract ectopic calcification in a mouse model for pseudoxanthoma elasticum. J. Mol. Med. 2011, 89, 1125.
- Fülöp, K.; Jiang, Q.; Wetering, K.V.; Pomozi, V.; Szabó, P.T.; Arányi, T.; Sarkadi, B.; Borst, P.; Uitto, J.; Váradi, A. ABCC6 does not transport vitamin K3-glutathione conjugate from the liver: Relevance to pathomechanisms of pseudoxanthoma elasticum. Biochem. Biophys. Res. Commun. 2011, 415, 468–471.
- Miglionico, R.; Armentano, M.F.; Carmosino, M.; Salvia, A.M.; Cuviello, F.; Bisaccia, F.; Ostuni, A. Dysregulation of gene expression in ABCC6 knockdown HepG2 cells. Cell. Mol. Biol. Lett. 2014, 19, 517–526.
- Ostuni, A.; Carmosino, M.; Miglionico, R.; Abruzzese, V.; Martinelli, F.; Russo, D.; Laurenzana, I.; Petillo, A.; Bisaccia, F. Inhibition of ABCC6 Transporter Modifies Cytoskeleton and Reduces Motility of HepG2 Cells via Purinergic Pathway. Cells 2020, 9, 1410.
- Miglionico, R.; Ostuni, A.; Armentano, M.F.; Milella, L.; Crescenzi, E.; Carmosino, M.; Bisaccia, F. ABCC6 knockdown in HepG2 cells induces a senescent-like cell phenotype. Cell. Mol. Biol. Lett. 2017, 22, 7.
- Pasquali-Ronchetti, I.; Garcia-Fernandez, M.I.; Boraldi, F.; Quaglino, D.; Gheduzzi, D.; Paolinelli, C.D.V.; Tiozzo, R.; Bergamini, S.; Ceccarelli, D.; Muscatello, U. Oxidative stress in fibroblasts from patients with pseudoxanthoma elasticum: Possible role in the pathogenesis of clinical manifestations. J. Pathol. J. Pathol. Soc. Great Br. Irel. 2006, 208, 54–61.
- Martinelli, F.; Cuviello, F.; Pace, M.C.; Armentano, M.F.; Miglionico, R.; Ostuni, A.; Bisaccia, F. Extracellular ATP regulates CD73 and ABCC6 expression in HepG2 Cells. Front. Mol. Biosci. 2018, 5, 75.
More