Poly (adenosine diphosphate-ribose) polymerase inhibitors (PARPis) belong to a class of targeted drugs developed for the treatment of homologous recombination repair (HRR)-defective tumors. Preclinical and limited clinical data suggest that PARP inhibition is effective against prostate cancer (PC) in patients with HRR-deficient tumors and that PARPis can improve the mortality rate of PC in patients with BRCA1/2 mutations through a synthetic lethality.
- PARP inhibitors
- synthetic lethality
- prostate cancer (PC)
- the latest research progress
- combination strategies
1. PARP Proteins
PARPs are nuclear enzymes that catalyze poly ADP ribosylation in most eukaryotic cells. They are involved in the posttranslational modification of various proteins. The PARP family includes 17 subtypes, including PARP1, PARP2, PARP3, and PARP7. PARP1 is the earliest and most intensively studied subtype of the PARP family. It is a rich ribozyme encoded by ADPRT1 (ADP ribozyme transferase-1 gene) at q41-42 on chromosome 1 and has an N-terminal zinc finger domain, namely, a DNA binding domain (DBD), a central self-modifying domain (AD), and a C-terminal catalytic domain (CAT) [1]. It participates in various functions, including DNA methylation; cell apoptosis; cell proliferation and differentiation; gene transcription regulation; chromatin remodeling; and, most importantly, DDR [2]. PARP1 is a DNA damage sensor and signal transducer that can recognize a wide range of damaged DNA structures including DSBs, single-strand breaks, gaps, and a range of non–B-form structures in which the base-stacking continuity of at least one strand is disrupted [3], and activates the catalytic process of PARP1. PARP1 catalyzes the decomposition of NAD+ into nicotinamide and ADP ribose, then takes ADP ribose as a substrate to make the receptor protein that includes PARP itself, together with histones and other chromatin-associated proteins, forming the poly (ADP-ribose) (PAR) chains [4][5]. This PARylation of proteins in the vicinity of the DNA breaks then likely mediates DNA repair by modifying chromatin structure (e.g., via histone-PARylation) and by localizing DNA repair effectors (e.g., XRCC1). PARP1 autoPARylation eventually leads to its own release from the site of DNA damage [6][7].
2. The Mechanism of Action of PARP Inhibitors
PARP1 is a key enzyme in SSB repair (SSBR), and the PARP inhibitors were initially developed as catalytic inhibitors to block SSB repair [7]. PARP1 has been strongly implicated in promoting SSBR [8]. Direct SSBs are detected by PARP1, which ribosylates itself and, most likely, other proteins in the vicinity of the break (e.g., histones). PARP1 activity results in recruitment/retention of the putative chromatin remodeling proteins APLF and ALC1, and importantly also XRCC1, a scaffold protein that promotes retention and/or activity of core SSBR enzymes including factors required to process damaged termini [9]. In cells, PARP1 inhibition causes failure of SSB repair. Furthermore, persistent SSBs have been shown to stall and collapse the replication fork, leading to DSB [9][10].HR-competent cells can repair DSBs for cell survival, whereas in cells with defects caused by mutations in breast cancer-associated antigens (BRCA1 or BRCA2) or other HR-associated proteins, the damage remains unrepaired, leading to cell death (
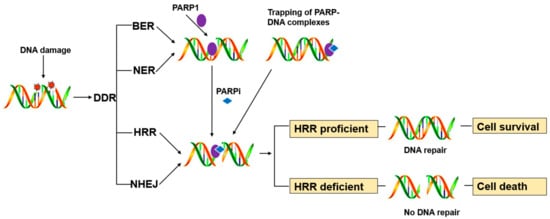
PARP inhibitors have also been reported to trap PARP enzymes in DNA. PARP inhibitors captured the PARP1 and PARP2 enzymes at the DNA damage sites, trapped PARP-DNA complexes were more cytotoxic than unrepaired SSBs caused by PARP inactivation, implying that PARP inhibitors act, in part, as toxins that trap PARP enzymes on DNA. The toxic PARP-DNA complex prevents replication fork progression and leads to cell death unless the damage is repaired. In addition, among the inhibitors, niraparib-treated cells were more capable of trapping than olaparib or veliparib-treated cells [12][13].
A third mechanism of PARP inhibitor sensitivity was discovered recently [14][15]. DSBs are dominantly repaired by HR in the S and G2 phase when sister chromatids are present [16]. In the presence of HR defects, tumor cells must rely on another microhomology-mediated end-joining or Alt-EJ pathway for repair. The pathway is dependent on PARP1 and translational ion polymerase (POLQ) [17]. Indeed, PARP1 is required to effectively recruit POLQ to DSBs. Therefore, inhibitors of PARP1 or POLQ can block the Alt-EJ pathway and kill HR-deficient tumor cells.