Nuclear factor-erythroid 2-related factor 2 (NRF2) and its major negative modulator Kelch-like ECH-associated protein 1 (KEAP1) are main players of the cellular defense mechanisms against internal and external cell stressors.
- Nrf2
- Keap1
- cancer
- oxidative stress
1. Introduction
Cancer is a non-communicable disease with an increasing incidence and mortality in many countries worldwide, which is expected to become the leading cause of death in every continent by the end of this century. According to the global statistical data published by (World Health Organization (WHO), Geneva, Switzerland) the main reason behind the growth of cancer-related deaths is the expansion of the world’s aging population [1]. Another critical phenomenon is that common cancer profiles have been changing in a way that infection and or poverty-related cancers tend to decrease while cancers that are associated with Westernized lifestyle tend to augment [2]. In this context, it is of great importance that researchers define the roles of critical molecular players related to tumor formation, progression, and metastasis. At the molecular level, cell division and death of damaged/mutated cells are tightly controlled by several pathways in order to prevent survival of damaged cells bearing mutations and pass those mutations to the next generations. Sometimes a critically damaged cell can take a life-changing decision and takes steps to secure its own survival despite the cost of transforming into a tumor cell. The steps to be taken in the way of transformation by a precancerous cell are described in the highly cited Weinberg review in detail [3]. Genes and signal transduction pathways common to more than one hallmark of cancer recently gained extra attention as promising therapeutic targets. Among the others, redox signaling emerged as a signal transduction pathway involved in every step of carcinogenesis [4]. Nuclear factor erythroid 2-related factor 2 (NRF2), also known as NFE2L2, is considered as the leading transcription factor controlling cellular redox homeostasis and antioxidant pathways [5]. Being the major stress regulator of the cell, NRF2 is involved in tumor formation, progression, and metastasis [6]. For this reason, a large number of studies is currently ongoing to better characterize NRF2 pathway and its roles in cancer. NRF2 displays a complex behavior in carcinogenesis [7]. To fully address the importance of NRF2 pathway in cancer, it is necessary to provide a description of its negative regulator KEAP1, that interacts with NRF2 to downmodulate its expression in cells and strictly control cellular homeostasis [8]. Under normal or low/moderate stress conditions, there is a tight balance between KEAP1 activity and NRF2 protein levels, which provides regulated antioxidant response, detoxification, and prevention of cancer. However, excessive stress, continuous overexpression of NRF2 or downregulation of KEAP1 cause a shift in this balance, which, in turn, acts in favor of carcinogenesis. Unraveling these roles would provide researchers to target NRF2 pathway in a more selective way to fully eradicate cancer without promoting its pro-oncogenic functions also known as the “dark side” of NRF2 [9]. In this respect, experimental studies wherein NRF2 function was abrogated with genetic or chemical approaches, support the notion that this transcription factor plays a cytoprotective role, acting as a tumor suppressor in specific contexts [10][11]. It has also been reported that NRF2 loss is strongly associated with tumor malignancy and metastatic behavior of cancer cells [12]. Moreover, partial or complete depletion of KEAP1 has been shown to promote cancer initiation and growth suggesting that KEAP1 can be also regarded as a tumor suppressor, similarly to NRF2 [13][14]. Based on collective data on NRF2 and KEAP1, there is a growing interest in better defining the therapeutic use of natural obtained or chemically synthesized activators of NRF2 with tumor-suppressing properties [15]. Yet, none of these compounds, either from natural or chemical sources, showed a consistent, stable, and dose-dependent effect to become a solid anticancer drug candidate, due to the context-dependent effects of NRF2 pathway in tumors [16]. Moreover, many studies reported that the abnormal activation of NRF2 is a common event in tumor cells, caused by several factors like somatic mutations, oncogenic signaling, epigenetic changes, metabolic reprograming and altered redox balance in cancer cells [17]. Indeed, abnormal NRF2 expression has been detected in various tumors such as lung, esophageal, laryngeal, skin, pancreas and liver cancers [18]. Despite these observations might argue against the assumption of NRF2 being a tumor suppressor, they actually indicate that NRF2, is a context-dependent transcription factor that can act as an oncogene under certain circumstances. In addition, abnormal KEAP1 expression has been observed in several cancers including lung, liver, pancreas, and ovarian cancers [19]. Thus, based on these data, it appears that there is a fine-tuning between KEAP1 and NRF2 levels and this determines which effect of this pathway will be more prominent under specific circumstances of a certain type of tumor. On the other hand, extensive research is focusing on NRF2 inhibitors in consideration of its cancer promoting roles, especially in the later stages of tumorigenesis [16]. However, these inhibitors may lack specificity and therefore interact with other downstream pathways causing undesired effects. Thus, it is of great importance that these drug candidates will be meticulously tested in large clinical studies in order to identify the specific cohorts of patients and the clinical context most likely having beneficial effects and minimal side toxicity.
Cancer is a non-communicable disease with an increasing incidence and mortality in many countries worldwide, which is expected to become the leading cause of death in every continent by the end of this century. According to the global statistical data published by (World Health Organization (WHO), Geneva, Switzerland) the main reason behind the growth of cancer-related deaths is the expansion of the world’s aging population [1]. Another critical phenomenon is that common cancer profiles have been changing in a way that infection and or poverty-related cancers tend to decrease while cancers that are associated with Westernized lifestyle tend to augment [2]. In this context, it is of great importance that researchers define the roles of critical molecular players related to tumor formation, progression, and metastasis. At the molecular level, cell division and death of damaged/mutated cells are tightly controlled by several pathways in order to prevent survival of damaged cells bearing mutations and pass those mutations to the next generations. Sometimes a critically damaged cell can take a life-changing decision and takes steps to secure its own survival despite the cost of transforming into a tumor cell. The steps to be taken in the way of transformation by a precancerous cell are described in the highly cited Weinberg review in detail [3]. Genes and signal transduction pathways common to more than one hallmark of cancer recently gained extra attention as promising therapeutic targets. Among the others, redox signaling emerged as a signal transduction pathway involved in every step of carcinogenesis [4]. Nuclear factor erythroid 2-related factor 2 (NRF2), also known as NFE2L2, is considered as the leading transcription factor controlling cellular redox homeostasis and antioxidant pathways [5]. Being the major stress regulator of the cell, NRF2 is involved in tumor formation, progression, and metastasis [6]. For this reason, a large number of studies is currently ongoing to better characterize NRF2 pathway and its roles in cancer. NRF2 displays a complex behavior in carcinogenesis [7]. To fully address the importance of NRF2 pathway in cancer, it is necessary to provide a description of its negative regulator KEAP1, that interacts with NRF2 to downmodulate its expression in cells and strictly control cellular homeostasis [8]. Under normal or low/moderate stress conditions, there is a tight balance between KEAP1 activity and NRF2 protein levels, which provides regulated antioxidant response, detoxification, and prevention of cancer. However, excessive stress, continuous overexpression of NRF2 or downregulation of KEAP1 cause a shift in this balance, which, in turn, acts in favor of carcinogenesis. Unraveling these roles would provide researchers to target NRF2 pathway in a more selective way to fully eradicate cancer without promoting its pro-oncogenic functions also known as the “dark side” of NRF2 [9]. In this respect, experimental studies wherein NRF2 function was abrogated with genetic or chemical approaches, support the notion that this transcription factor plays a cytoprotective role, acting as a tumor suppressor in specific contexts [10,11]. It has also been reported that NRF2 loss is strongly associated with tumor malignancy and metastatic behavior of cancer cells [12]. Moreover, partial or complete depletion of KEAP1 has been shown to promote cancer initiation and growth suggesting that KEAP1 can be also regarded as a tumor suppressor, similarly to NRF2 [13,14]. Based on collective data on NRF2 and KEAP1, there is a growing interest in better defining the therapeutic use of natural obtained or chemically synthesized activators of NRF2 with tumor-suppressing properties [15]. Yet, none of these compounds, either from natural or chemical sources, showed a consistent, stable, and dose-dependent effect to become a solid anticancer drug candidate, due to the context-dependent effects of NRF2 pathway in tumors [16]. Moreover, many studies reported that the abnormal activation of NRF2 is a common event in tumor cells, caused by several factors like somatic mutations, oncogenic signaling, epigenetic changes, metabolic reprograming and altered redox balance in cancer cells [17]. Indeed, abnormal NRF2 expression has been detected in various tumors such as lung, esophageal, laryngeal, skin, pancreas and liver cancers [18]. Despite these observations might argue against the assumption of NRF2 being a tumor suppressor, they actually indicate that NRF2, is a context-dependent transcription factor that can act as an oncogene under certain circumstances. In addition, abnormal KEAP1 expression has been observed in several cancers including lung, liver, pancreas, and ovarian cancers [19]. Thus, based on these data, it appears that there is a fine-tuning between KEAP1 and NRF2 levels and this determines which effect of this pathway will be more prominent under specific circumstances of a certain type of tumor. On the other hand, extensive research is focusing on NRF2 inhibitors in consideration of its cancer promoting roles, especially in the later stages of tumorigenesis [16]. However, these inhibitors may lack specificity and therefore interact with other downstream pathways causing undesired effects. Thus, it is of great importance that these drug candidates will be meticulously tested in large clinical studies in order to identify the specific cohorts of patients and the clinical context most likely having beneficial effects and minimal side toxicity.
2. NRF2 and KEAP1 Signaling Pathway
NRF2 was discovered in 1994 and belongs to the Cap and Collar (CNC) basic-region leucine zipper transcription factor family [20]. NRF2 has seven conserved NRF2-ECH homology domains comprising Neh1 to Neh7 (A). Neh1, Neh3, Neh4, and Neh5 domains are involved in the transcriptional activation of NRF2 by binding its co-activators. Neh2, Neh6, and Neh7 control the stability of NRF2 through responding as a negative regulatory domain [16]. Neh1 domain is known as a CNC-bZIP domain that allows NRF2 to bind antioxidant response element (ARE), also known as the electrophile response element (EpRE) through interaction with other factors like small musculoaponeurotic fibrosarcoma (sMAF) [21]. Neh2 domain functions as a major regulatory domain of NRF2 containing ETGE and DLG regions that are required for the interaction with KEAP1. In addition, Neh2 domain has lysine rich residues responsible for the ubiquitination and subsequent proteasomal degradation of NRF2 [22]. Neh3 is the transactivation domain recruiting co-activators that are necessary for the transactivation of NRF2 [23]. NRF2 also possesses Neh4 and Neh5 domains containing acid-rich residues that interact with CREB-binding protein with histone acetyltransferase activity (CBP) [24]. The Neh6 domain contains serine-rich residues that can be phosphorylated by Glycogen Synthase Kinase 3b (GSK-3β) and leads to proteasomal degradation of NRF2 through cullin 1 (Cul1)-dependent ubiquitination [25]. The Neh7 domain mediates the binding of RXRα (retinoid X receptor α) that inhibits the NRF2 transcriptional activity [26].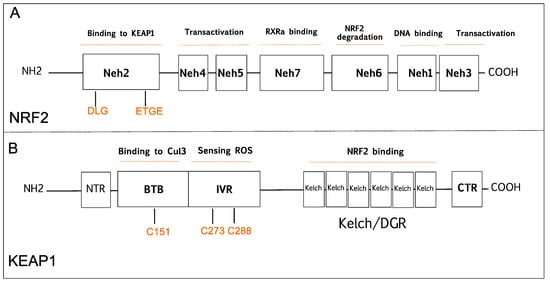
Figure 1.
A
B
2S) [29][30][31]. The Kelch/DGR domain functions as an NRF2 repressor, and it contains of six Kelch motif repeats, which are required for interaction with Neh2 domain of NRF2 [32].
S) [29,30,31]. The Kelch/DGR domain functions as an NRF2 repressor, and it contains of six Kelch motif repeats, which are required for interaction with Neh2 domain of NRF2 [32]. NRF2 increases cellular antioxidant capacity by controlling the expression of detoxifying and antioxidant genes. Hence, NRF2 has been previously known as a transcription factor that inhibits cancer development. Under homeostatic conditions, KEAP1 plays a critical role in NRF2 activity by binding to DLG/ETGE motifs in the Neh2 domain, and keeps NRF2 protein at low levels in the cytoplasm by promoting its polyubiquitylation and proteasomal degradation [33]. However, under stress conditions, highly reactive cysteine residues in KEAP1 are oxidized, and this modification disrupts the binding of KEAP1 to NRF2 promoting its nuclear translocation, wherein NRF2 forms NRF2-sMaf heterodimers via its Neh1 domain and induces gene expression by binding to the ARE sequences in the promoter regions of NRF2 target genes [33]. KEAP1-NRF2 pathway is one of the major signaling cascades that promote antioxidant defense in normal cells, which is a crucial mechanism in the prevention of cancer development. Many studies have shown that KEAP1 and NRF2 proteins function as tumor suppressors, as their absence leads to tumorigenesis while other work indicates that NRF2 can also promote tumor progression. In the following sections, we will briefly discuss past and present studies focused on this seemingly paradoxical aspect.2.1. The Tumor Suppressive Role of NRF2 Pathway
A strong indication supporting a tumor-suppressive role of the NRF2 signaling derives from a number of in vivo studies comparing the sensitivity to chemically induced carcinogenesis in NRF2-knockout mice (Nfe2l2-/-) and wild-type mice. In this respect, it was found that NRF2-null mice are more susceptible to developing bladder, skin, and stomach cancer when exposed to chemical carcinogens compared to wild-type mice [34]. In addition, the basal expression level of ARE-mediated genes such as GCL, GST, HMOX1, NQO1, and UGT was found to be significantly suppressed in NRF2-deficient mice compared to the wild-type counterpart [11][35][36]. The mechanism, by which NRF2 protects cells from chemical-induced carcinogenesis, appears to depend on its role in detoxification of chemical carcinogens and ROS, and the induction of DNA damage repair mechanisms that ultimately prevent mutations. In a study with mice harboring SNPs (single nucleotide polymorphisms) in the promoter region of the NRF2 gene, it was shown that reduced NRF2 expression made mice more susceptible to lung injury due to hypoxia [37]. SNP-bearing individuals have lower NRF2 mRNA levels causing an elevated risk of developing non-small cell lung cancer (NSCLC) [13][14].
2.2. The Tumor Suppressive Role of KEAP1
2.3. The Carcinogenic Role of NRF2
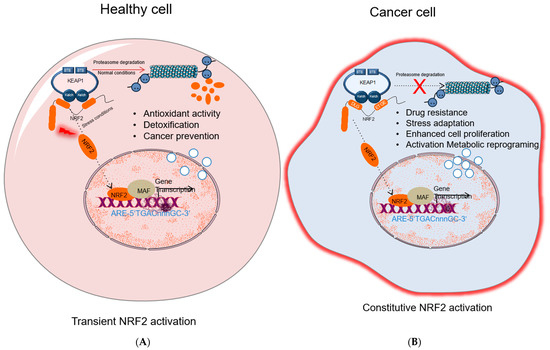
Figure 2.
A
B
Moreover, persistent activation of NRF2 was reported to attenuate the toxicity of ionizing radiation and drug treatment in human lung cancer cells, while NRF2 knockdown enhanced cellular response to ionizing radiation and chemotherapeutic drugs. These findings suggest that targeting NRF2 activity alone or in combination with other drugs could be an effective strategy to improve the sensitivity of malignant cells to anticancer therapies [44][45].
2.4. The Carcinogenic Role of KEAP1
Despite protective effects on cancer progression, studies have also demonstrated the carcinogenic role of KEAP1 mutations in various cancers such as gallbladder, prostate, liver, colorectal, lung, breast, and prostate cancers [51][52][53][54]. Some mutations found in the N-terminal and BTB domains of KEAP1 prevented ubiquitination of NRF2 through the disruption of KEAP1-CUL3 formation, and other mutations in the Kelch domains inhibited interaction of KEAP1-NRF2 and caused stabilization of NRF2 [45][46][55]. Additionally, mutations in KEAP1 were also detected in liver and gallbladder, which caused over expression of antioxidant and phase II detoxification enzymes that have roles in cancer chemo-resistance [53][56].
3. NRF2 Activation Mechanisms in Cancer
Comprehensive studies validated that NRF2-KEAP1 signaling pathway is activated in several cancers such as skin, lung, bladder, hepatocellular carcinoma, esophagus, ovarian, prostate, pancreatic, and breast cancer [6][52][53][55][57]. The molecular mechanisms responsible for the activation of NRF2 in cancer are schematized in
Comprehensive studies validated that NRF2-KEAP1 signaling pathway is activated in several cancers such as skin, lung, bladder, hepatocellular carcinoma, esophagus, ovarian, prostate, pancreatic, and breast cancer [6,52,53,55,57]. The molecular mechanisms responsible for the activation of NRF2 in cancer are schematized in
and further details are discussed below:
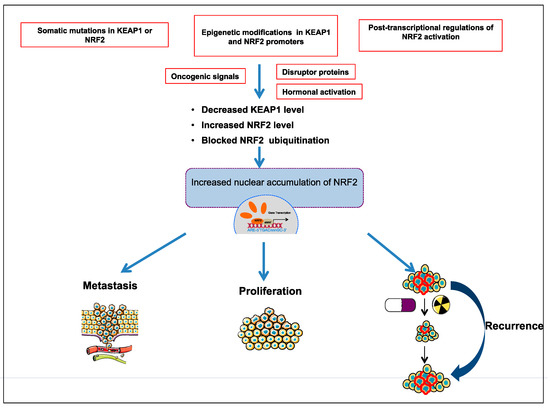
Figure 3.
3.1. The Somatic Mutations in KEAP1 or NRF2
KEAP1
NFE2L2 genes are the most known mechanisms that reduce NRF2-KEAP1 binding and prevent degradation of NRF2 through KEAP1/CUL3/RBX1 E3-ubiquitin ligase complex [45][51]. Increasing evidence has established that the inhibition of NRF2-KEAP1 interaction leads to the overexpression of NRF2 in cancer cells that, in turn, enhances the activation of antioxidant defense system, and proteins involved in chemoresistance and radioresistance system via activating ARE-containing gene expression. Most of the inactivating mutations in the
NFE2L2
NF2EL2
KEAP1
NFE2L2
KEAP1 mutations can be missense or nonsense mutations and observed on the entire gene [45][55]. Some of the mutations in
KEAP1 gene lead to deregulation of apoptosis, autophagy, and inflammation by accumulation of BCL2 and p62 proteins [59][60]. The first loss-of-function mutations in Kelch/DGR domain of KEAP1 were reported in human lung adenocarcinoma cell lines [54]. Then, somatic mutations in Kelch/IVR domain of KEAP1 were detected in both human NSCLC cell lines and clinical NSCLC patients’ tumor samples [45][55]. Recently, different research groups also reported that
KEAP1 genetic alterations could be novel molecular hallmarks in high neuroendocrine gene expressing lung cancers [61][62].
3.2. Epigenetic Modifications in KEAP1 and NRF2 Promoters
KEAP1
NFE2L2
KEAP1
KEAP1 promoter was found to be significantly hypermethylated [43][63][64][65]. Moreover, hypermethylation within the promoter region of
KEAP1
NFE2L2
KEAP1
NFE2L2
3.3. Post-Transcriptional Regulation of NRF2 Activation
KEAP1 mRNA expression and induce NRF2 activation [70]. It was reported that miR-141 is overexpressed in breast and ovarian cancer, and additionally, overexpression of this miRNA increased chemoresistance of HCC cells to 5-fluorouracil through the activation of NRF2-driven antioxidant pathways [71][72].
3.4. Disruptor Proteins
Several disrupting proteins are involved in the activation of NRF2 in cancer. Moreover, p62, also known as sequestosome 1 (SQSTM1), is an autophagy receptor protein that contains the STGE motif, which is similar to the ETGE motif of NRF2. This protein competes with NRF2 for KEAP1 binding and promotes autophagic degradation of KEAP1 [73][74][75][76]. Studies proved that when p62 expression was decreased by siRNA-mediated knockdown, NRF2 and its target genes were downregulated, while the half-life of KEAP1 increased by twofold [73][76]. In addition, elevated p62 contributed to renal cancer progression and hepatocellular carcinoma through the activation of NRF2 [77][78][79]. These studies emphasize the critical role of p62 and NRF2 axis in the regulation of tumor development.
Besides, p21, which is a direct target of p53, associates with ETGE and/or DLG motifs in NRF2 and disrupts NRF2-KEAP1 binding causing NRF2 accumulation [80]. Furthermore, Wilms tumor gene on the X chromosome (WTX) and partner and localizer of BRCA2, also known as PALB2 proteins have been shown to bind KEAP1 and suppress NRF2 ubiquitination [81][82]. Similarly, the protein dipeptidyl peptidase 3 (DPP3) was shown to inhibit NRF2 ubiquitination through binding to KEAP1, thus activating NRF2-dependent gene transcription in breast cancer [83].
3.5. Oncogenic Signals
KRAS
BRAF
C-MYC
3.6. Hormonal Activation
Several studies validated the effects of hormonal activation of NRF2 on cancer progression. Gonadotropins and sex steroid hormones, including follicle-stimulating hormone (FSH), estrogen (E2), and luteinizing hormone (LH), have been reported to be critical in activation of NRF2 through the induction of ROS that inhibit KEAP1 by oxidation of its cysteine residues [88]. In addition, follicle-stimulating hormone (FSH) is known to induce expression of vascular endothelial growth factor (VEGF) and hypoxia inducible factor 1α (HIF1α). Thus, FSH contributes to tumor angiogenesis through ROS-mediated NRF2 signaling [89].