In humans, the glutathione S-transferases (GST) protein family is composed of seven members that present remarkable structural similarity and some degree of overlapping functionalities. GST proteins are crucial antioxidant enzymes that regulate stress-induced signaling pathways. Interestingly, overactive GST proteins are a frequent feature of many human cancers. Recent evidence has revealed that the biology of most GST proteins is complex and multifaceted and that these proteins actively participate in tumorigenic processes such as cell survival, cell proliferation, and drug resistance. Structural and pharmacological studies have identified various GST inhibitors, and these molecules have progressed to clinical trials for the treatment of cancer and other diseases.
- Glutathione S-transferases (GSTs)
- antioxidants
- cancer-cell signaling
- chemoresistance
1. Introduction
Glutathione S-transferases (GSTs) are a multigene family (EC 2.5.1.18) of eight dimeric enzymes that are classified based on their amino acid sequences and substrate specificity as alpha (A), kappa (K), mu (M), omega (O), pi (P), sigma (S), theta (T), and zeta (Z) [1]. Depending on their subcellular location, GSTs are grouped as cytoplasmic (A, P, M, S, T, Z), mitochondrial (K), or membrane-bound (Membrane Associated Proteins in Eicosanoid and Glutathione metabolism) [2]. GSTs are phase-II detoxification enzymes found in most life forms and vital for maintaining cellular homeostasis [3]. GSTs play a cytoprotective role primarily by catalyzing the conjugation reaction of reduced glutathione (GSH) and reactive electrophiles generated by cytochrome P450 metabolism to form GSH conjugates [4]. The resulting GSH conjugates are either excreted via bile or transported to the kidney where: (1) the γ-glutamyl moiety is cleaved by γ-glutamyl transpeptidase; (2) the glycine is cleaved by dipeptidase; and (3) the cysteine is N-acetylated [5].
In addition to their detoxification roles, GSTs are known for their functions in cell signaling, post-translational modification, and resistance to chemotherapeutic agents [6]. For example, the pi and mu classes of GSTs regulate the mitogen-activated protein (MAP) kinase pathway that governs cell survival and cell death signals via direct interactions with c-Jun N-terminal kinase 1 (JNK1) and apoptosis signal-regulating kinase (ASK1) [7]. Additionally, GSTs form complexes with an array of intracellular proteins for their post-translational modification [8]. For instance, protein disulfide isomerase (PDI), peroxiredoxin-VI (Prdx VI), and p53 are common substrates of GST-mediated glutathionylation [9]. Similar to the detoxification process described above, antineoplastic drugs bound to GSH are expelled out of the cells by the membrane-bound GS-X pump, making cancer cells resistant to chemotherapy [10]. Since their discovery in 1961 in rat liver [11], GSTs have gained attention among cancer researchers. The expression of GSTs in all cell types and their abundance in aggressive cancer cells suggest that they play a key role in tumor progression and cancer pathogenicity [12]. Recent developments in the field of redox oncology have shed light on novel functions of GST proteins in cancer cells [13][14]. This review summarizes newly identified functions of GST proteins and their roles in the cellular signaling, metabolism, and survival of cancer cells.
2. Structure
Because GSTs are pivotal in drug metabolism, they were among the first cytosolic proteins to be structurally characterized. Porcine GST Pi 1 (GSTP1)was the first member of the family whose structure was determined [15]. Crystallographic studies have revealed that the catalytic GSTs display analogous tertiary structures and exist as homodimers in mammals (
A) [16]; however, heterodimers of a few cytosolic GSTs have been identified in plants [17]. Currently, no enzymatically active monomers of GST proteins are known [15]. Subsequent structural analyses revealed that all principal GST family members have a basic protein fold consisting of two domains: the N-terminal domain and the C-terminal domain. The GST N-terminal domain fold is similar to other cellular homeostasis and detoxification proteins such as glutathione peroxidases and glutaredoxins. The N-terminal domain constitutes approximately one-third of the protein structure and is made up of a β-α-β-α-β-β-α motif. The β-β-α motif in the N-terminal domain, also known as the G-site, is most conserved among the isoforms and provides the binding site for reduced glutathione (GSH) by recognizing the γ-glutamyl fragment of GSH (
B).
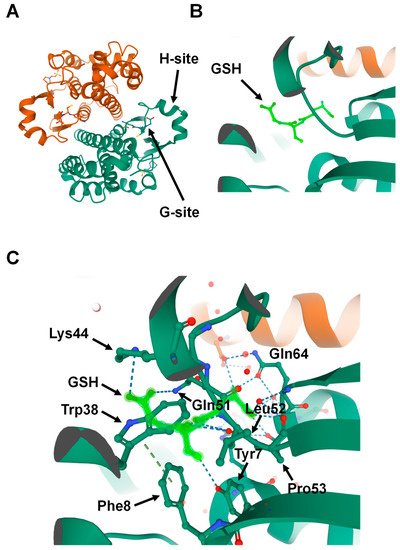
Structure of Glutathione S-transferase Pi 1 (GSTP1) (Protein Data Bank ID: 6GSS). (
) Homodimer assembly of GSTP1 showing G- and H-sites. (
) Magnified view of the G-site that is occupied by the ligand glutathione (GSH) (shown in light green). (
) Glutathione (light green) forms hydrogen-binding interactions with the surrounding amino acids found in the G-site pocket of GSTP1.
Interestingly, a proline residue, found at the N-terminal end of strand β3, is conserved among all cytosolic and mitochondrial GSTs. This proline forms hydrogen-bond interactions with the backbone amine group of the GSH-cysteinyl moiety (
C) [16][18][19]. Global characterization of sequence and structure similarity of GST proteins show two major subgroups: (1) tyrosine-type GSTs (Y-GSTs), which use tyrosine to activate GSH; and (2) S/C-GSTs, which use serine (or cysteine in case of GST Omega (GSTO)) to interact with GSH [20]. However, the C-terminal domain of GSTs constitutes the other two-thirds of the protein structure and is made up of a unique all-α-helical domain [19]. The hydrophobic substrates bind to a cleft between the N- and C-terminal domains known as the H-site. Unlike the G-site, the H-site is highly variable in shape and chemical constitution between classes [21]. This variability in H-site structure determines the substrate selectivity of various GST isozymes [22].
3. Conclusions and Future Directions
GST proteins have complex biology and play multifaceted roles in cancer cells. These enzymes are a crucial component of the cellular antioxidant system and play critical roles in maintaining cellular homeostasis. Under normal physiological conditions, GSTP1 can glutathionylate multiple proteins, including various transcription factors and oncogenes. Conversely, under oxidative stress, GSTP1 can trigger MAPK- and caspase-mediated apoptotic signaling pathways. Interestingly, recent findings suggest that GST enzymes play important roles in cancer development and chemoresistance. However, kinetic and functional studies have revealed that most antineoplastic agents are poor substrates of GSTs with a weaker catalytic constant for the conjugation reaction. Therefore, researchers have shifted their focus to investigating the role of GSTs in various cellular functions, such as regulating kinases and the post-translational processes of diverse proteins.
Multiple studies have demonstrated that GST proteins are overexpressed in many human cancers. Their overexpression contributes to poor outcomes and is negatively correlated with patient survival. However, GSTP1 is not considered a diagnostic marker in clinical practices. We suggest that GSTP1, along with a combination of other biomarkers, may identify a high-risk population that is susceptible to developing cancer. In conclusion, recent studies have established the role of GSTP1 and other GST isozymes in cancer development, progression, metastasis, and resistance to antineoplastic drugs. Active research in the field of antioxidants and redox biology has narrowed to GSTP1 as a promising therapeutic target for cancer treatment. GSTP1 inhibitors can potentially be used in the future to enhance the efficacy of chemotherapy and overcoming drug resistance. However, to use these inhibitors safely for cancer treatment, research is needed to characterize their impact on normal cells and the long-term effects.
References
- Hayes, J.D.; Flanagan, J.U.; Jowsey, I.R. Glutathione transferases. Annu. Rev. Pharmacol. Toxicol. 2005, 45, 51–88.
- Chatterjee, A.; Gupta, S. The multifaceted role of glutathione S-transferases in cancer. Cancer Lett. 2018, 433, 33–42.
- Zhang, J.; Grek, C.L.; Ye, Z.-W.; Manevich, Y.; Tew, K.D.; Townsend, D.M. Pleiotropic Functions of glutathione S-transferase P. Adv. Cancer Res. 2014, 122, 143–175.
- Kural, C.; Kocdogan, A.K.; Şimşek, G.G.; Oguztuzun, S.; Kaygın, P.; Yılmaz, I.; Bayram, T.; Izci, Y. Glutathione S-transferases and cytochrome P450 enzyme expression in patients with intracranial tumors: Preliminary report of 55 patients. Med. Princ. Pr. 2018, 28, 56–62.
- Soleo, L.; Strzelczyk, R. Xenobiotics and glutathione. G Ital. Med. Lav. Ergon. 1999, 21, 302–308.
- Tew, K.D.; Townsend, D.M. Regulatory functions of glutathioneS-transferase P1-1 unrelated to detoxification. Drug Metab. Rev. 2011, 43, 179–193.
- Adler, V.; Yin, Z.; Fuchs, S.Y.; Benezra, M.; Rosario, L.; Tew, K.D.; Pincus, M.R.; Sardana, M.; Henderson, C.J.; Wolf, C.; et al. Regulation of JNK signaling by GSTp. EMBO J. 1999, 18, 1321–1334.
- Fratelli, M.; Demol, H.; Puype, M.; Casagrande, S.; Eberini, I.; Salmona, M.; Bonetto, V.; Mengozzi, M.; Duffieux, F.; Miclet, E.; et al. Identification by redox proteomics of glutathionylated proteins in oxidatively stressed human T lym-phocytes. Proc. Natl. Acad. Sci. USA 2002, 99, 3505–3510.
- Townsend, D.M.; Manevich, Y.; He, L.; Hutchens, S.; Pazoles, C.J.; Tew, K.D. Novel role for glutathione S-transferase pi. Regulator of protein S-Glutathionylation following oxidative and nitrosative stress. J. Biol. Chem. 2009, 284, 436–445.
- Júnior, P.I.D.B.; Senna, S.M.; Vidor, A.C.; Miyasaka, C.K.; Curi, R.; Williams, J.F. Glutathione metabolism and glutathione S-conjugate export ATPase (MRP1/GS-X pump) activity in cancer. II. Cell-to-cell variability, relation with cellular activation state and functional absence of GS-X pump in lymphocytes. Biochem. Mol. Boil. Int. 1998, 45, 1243–1254.
- Booth, J.; Boyland, E.; Sims, P. An enzyme from rat liver catalysing conjugations with glutathione. Biochem. J. 1961, 79, 516–524.
- Louie, S.M.; Grossman, E.A.; Crawford, L.A.; Ding, L.; Camarda, R.; Huffman, T.R.; Miyamoto, D.K.; Goga, A.; Weerapana, E.; Nomura, D.K. GSTP1 is a driver of triple-negative breast cancer cell metabolism and pathogenicity. Cell Chem. Biol. 2016, 23, 567–578.
- Checa-Rojas, A.; Delgadillo-Silva, L.F.; Velasco-Herrera, M.D.C.; Andrade-Domínguez, A.; Gil, J.; Santillán, O.; Lozano, L.; Toledo-Leyva, A.; Ramírez-Torres, A.; Talamas-Rohana, P.; et al. GSTM3 and GSTP1: Novel players driving tumor progression in cervical cancer. Oncotarget 2018, 9, 21696–21714.
- Townsend, D.M.; Findlay, V.J.; Fazilev, F.; Ogle, M.; Fraser, J.; Saavedra, J.E.; Ji, X.; Keefer, L.K.; Tew, K.D. A glutathione S-transferase pi-activated prodrug causes kinase activation concurrent with S-glutathionylation of proteins. Mol. Pharmacol. 2006, 69, 501–508.
- Reinemer, P.; Dirr, H.W.; Ladenstein, R.; Schäffer, J.; Gallay, O.; Huber, R. The three-dimensional structure of class pi glutathione S-transferase in complex with glutathione sulfonate at 2.3 A resolution. EMBO J. 1991, 10, 1997–2005.
- Oakley, A. Glutathione transferases: A structural perspective. Drug Metab. Rev. 2011, 43, 138–151.
- Kilili, K.G.; Atanassova, N.; Vardanyan, A.; Clatot, N.; Al-Sabarna, K.; Kanellopoulos, P.N.; Makris, A.M.; Kampranis, S.C. Differential roles of tau class glutathione S-transferases in oxidative stress. J. Biol. Chem. 2004, 279, 24540–24551.
- Ladner, J.E.; Parsons, J.F.; Rife, C.L.; Gilliland, G.L.; Armstrong, R.N. Parallel evolutionary pathways for glutathione transferases: Structure and mechanism of the mitochon-drial class kappa enzyme rGSTK1-1. Biochemistry 2004, 43, 352–361.
- Li, J.; Xia, Z.; Ding, J. Thioredoxin-like domain of human kappa class glutathione transferase reveals sequence homology and structure similarity to the theta class enzyme. Protein Sci. 2005, 14, 2361–2369.
- Atkinson, H.J.; Babbitt, P.C. Glutathione transferases are structural and functional outliers in the thioredoxin fold. Biochemistry 2009, 48, 11108–11116.
- Board, P.G.; Coggan, M.; Chelvanayagam, G.; Easteal, S.; Jermiin, L.S.; Schulte, G.K.; Danley, D.E.; Hoth, L.R.; Griffor, M.C.; Kamath, A.V.; et al. Identification, characterization, and crystal structure of the omega class glutathione transferases. J. Biol. Chem. 2000, 275, 24798–24806.
- Wilce, M.C.; Parker, M.W. Structure and function of glutathione S-transferases. Biochim. Biophys. Acta BBA Protein Struct. Mol. Enzym. 1994, 1205, 1–18.