Adipose tissue (AT) storage capacity is central in the maintenance of whole-body homeostasis, especially in obesity states. However, sustained nutrients overflow may dysregulate this function resulting in adipocytes hypertrophy, AT hypoxia, inflammation and oxidative stress. Other factors such as systemic inflammation and lifestyle behaviours may also contribute to the disruption of AT redox equilibrium and exacerbate obesity-associated oxidative stress.
- obesity
- oxidative stress
- adipose tissue
- metabolic dysfunctions
1. Introduction
Impaired AT expandability, potentially leading to AT dysfunction in obesity is an accepted theory for the development of obesity-associated metabolic disorders in some, but not all individuals [1]. AT dysfunction may be initiated by pathological mechanisms such as hypoxia that propagate several types of AT stresses including inflammatory, metabolic, endoplasmic reticulum and oxidative stress [1][2]. Oxidative stress has been demonstrated in individuals with obesity and is reflected by elevated markers of reactive oxygen species (ROS) such as isoprostanes, circulating thiobarbituric reactive acid substances (TBARS) or malondialdehyde (MDA), lipid hydroperoxides (LOOH), as well as reduced antioxidant defense system [3][4][5]. Oxidative stress has also been associated with systemic inflammation [3][6], impairment of glucose uptake into adipocytes [7], and decreased insulin secretion from pancreatic β-cells [8], supporting its critical role in the pathogenesis of T2D. Besides, increased oxidative stress in accumulating AT has been associated with dysregulation of adipokine secretion and insulin resistance (IR) in individuals with obesity, while both increased systemic and AT oxidative stress have been associated with obesity-induced inflammation [3][9]. Therefore, understanding the relationship between obesity-induced oxidative stress and the associated metabolic disorders is of relevance for the elucidation of disease mechanisms and identification of treatment targets (
Impaired AT expandability, potentially leading to AT dysfunction in obesity is an accepted theory for the development of obesity-associated metabolic disorders in some, but not all individuals [10]. AT dysfunction may be initiated by pathological mechanisms such as hypoxia that propagate several types of AT stresses including inflammatory, metabolic, endoplasmic reticulum and oxidative stress [10,11]. Oxidative stress has been demonstrated in individuals with obesity and is reflected by elevated markers of reactive oxygen species (ROS) such as isoprostanes, circulating thiobarbituric reactive acid substances (TBARS) or malondialdehyde (MDA), lipid hydroperoxides (LOOH), as well as reduced antioxidant defense system [12,13,14]. Oxidative stress has also been associated with systemic inflammation [12,15], impairment of glucose uptake into adipocytes [16], and decreased insulin secretion from pancreatic β-cells [17], supporting its critical role in the pathogenesis of T2D. Besides, increased oxidative stress in accumulating AT has been associated with dysregulation of adipokine secretion and insulin resistance (IR) in individuals with obesity, while both increased systemic and AT oxidative stress have been associated with obesity-induced inflammation [12,18]. Therefore, understanding the relationship between obesity-induced oxidative stress and the associated metabolic disorders is of relevance for the elucidation of disease mechanisms and identification of treatment targets (
).
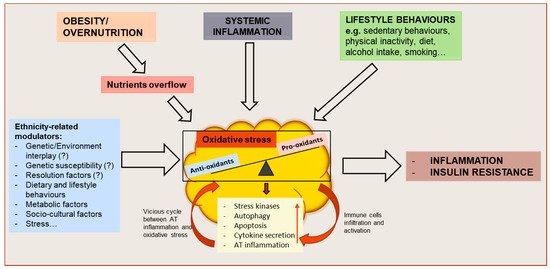
Figure 1.
Schematic representation of oxidative stress drivers and metabolic consequences on adipose tissue (AT) function and whole-body metabolism. Obesity or overnutrition may result in nutrients overflow to AT, resulting in adipocyte hypertrophy and AT hypoxia which might induce an oxidative stress state in the tissue. Systemic inflammation, as well as behavioral factors, may also contribute to the disruption of the redox equilibrium of AT. As a result, the activation of stress signaling pathways contribute to increasing autophagy and apoptosis, dysregulated adipokine secretion and AT inflammation. The resulting functional alterations may further impair AT function by causing an increased attraction, infiltration and activation of immune cells and increased AT inflammation, creating a vicious cycle between AT oxidative stress and inflammation, and leading to whole-body metabolic dysfunction. These mechanisms might be influenced by ethnicity-related modulators.
2. Oxidative Stress
Oxidative stress generally refers to an imbalance between the production of pro-oxidant substances (i.e., free radicals, ROS and/or reactive nitrogen species (RNS)) and the antioxidant defense system [3]. ROS are generated during cellular metabolism when the chemical reduction of oxygen forms unstable free radicals, characterized by an unpaired electron (e.g., superoxide (O
Oxidative stress generally refers to an imbalance between the production of pro-oxidant substances (i.e., free radicals, ROS and/or reactive nitrogen species (RNS)) and the antioxidant defense system [12]. ROS are generated during cellular metabolism when the chemical reduction of oxygen forms unstable free radicals, characterized by an unpaired electron (e.g., superoxide (O
2−
), hydrogen peroxide (H
2
O
2
), hydroxyl radical (OH
−
);
) [36].
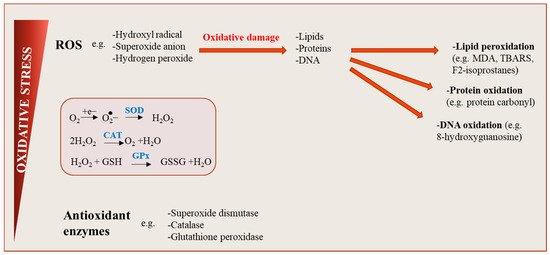
Figure 2.
Oxidative stress: an imbalance between reactive oxygen species (ROS) production and antioxidant defenses. ROS are generated during cellular metabolism when the chemical reduction of oxygen forms unstable free radicals. Several molecule types including lipids, proteins or nucleic acids can be oxidized or nitrated, and the resultant product, when accumulated in cells over time become harmful, affecting cell signaling pathways and tissue function. Physiological levels of ROS are conserved by the action of antioxidants, maintaining a redox balance in cells.
3. Oxidative Stress in Obesity
Given the major health burden caused by the double pandemic of obesity and T2D, it is important to understand the causal mechanisms underlying their relationship, as well as the implication of oxidative stress in this association. Oxidative stress has been evidenced during the development of obesity with elevated levels (urinary, systemic and/or tissue-specific) of biomarkers such as 8-epi-Prostaglandin F2α (8-iso-PGF2α), 4-Hydroxynonenal (4-HNE) and MDA in children and adults [11][12], insulin-sensitive and insulin-resistant patients with obesity [13][14][15][16][17][18]. Moreover, dysregulation in antioxidant defense (decrease or increase in antioxidants capacity) has been shown, as well as inverse associations between antioxidant capacity and body fat percentage. This suggests that the degree of adiposity affects antioxidant enzyme activities [18][19], and/or vice versa [20][21].
Given the major health burden caused by the double pandemic of obesity and T2D, it is important to understand the causal mechanisms underlying their relationship, as well as the implication of oxidative stress in this association. Oxidative stress has been evidenced during the development of obesity with elevated levels (urinary, systemic and/or tissue-specific) of biomarkers such as 8-epi-Prostaglandin F2α (8-iso-PGF2α), 4-Hydroxynonenal (4-HNE) and MDA in children and adults [50,51], insulin-sensitive and insulin-resistant patients with obesity [52,53,54,55,56,57]. Moreover, dysregulation in antioxidant defense (decrease or increase in antioxidants capacity) has been shown, as well as inverse associations between antioxidant capacity and body fat percentage. This suggests that the degree of adiposity affects antioxidant enzyme activities [57,58], and/or vice versa [59,60].
The increase of antioxidant enzymes in response to high ROS concentrations might be a compensatory response during early-stages of obesity development, in order to maintain the oxidative balance until the antioxidant capacity is depleted [18]. A sustained increase in the endogenous activity of antioxidant enzymes can reduce the incidence of oxidative stress and associated metabolic disorders by regulating ROS production [22][23]. For instance, antioxidant treatment improves insulin function in people living with diabetes [24]. In contrast, individuals with a genetic predisposition for low catalase activity (e.g., acatalasemia, catalase mutations) are at higher risk of T2D [25]. Moreover, the deletion of endogenous catalase (
The increase of antioxidant enzymes in response to high ROS concentrations might be a compensatory response during early-stages of obesity development, in order to maintain the oxidative balance until the antioxidant capacity is depleted [57]. A sustained increase in the endogenous activity of antioxidant enzymes can reduce the incidence of oxidative stress and associated metabolic disorders by regulating ROS production [62,63]. For instance, antioxidant treatment improves insulin function in people living with diabetes [64]. In contrast, individuals with a genetic predisposition for low catalase activity (e.g., acatalasemia, catalase mutations) are at higher risk of T2D [65]. Moreover, the deletion of endogenous catalase (
Cat−/− mice) results in the development of liver steatosis and inflammation both on chow and high-fat diets [20][26]. Furthermore,
mice) results in the development of liver steatosis and inflammation both on chow and high-fat diets [59,66]. Furthermore,
Cat−/− mice show a pre-diabetic phenotype characterized by impaired glucose tolerance and increased fasting serum insulin [20]. These and several other studies provide evidence for tight relationships between oxidative stress, central obesity and excess fat accumulation, as well as associated metabolic disorders such as inflammation, IR and T2D [9][11][27][28][29][30][31][32].
mice show a pre-diabetic phenotype characterized by impaired glucose tolerance and increased fasting serum insulin [59]. These and several other studies provide evidence for tight relationships between oxidative stress, central obesity and excess fat accumulation, as well as associated metabolic disorders such as inflammation, IR and T2D [18,50,67,68,69,70,71,72].
The occurrence of oxidative stress in obesity is further demonstrated by an upregulated expression of the ROS-producing enzyme NADPH (nicotinamide adenine dinucleotide phosphate) oxidase (NOX) in AT of patients with obesity and IR [9][33][34][35]. Notably, oxidative stress in AT may be distinct for specific fat depots. For instance, higher concentrations of hydrogen peroxide, catalase and SOD activities were shown in VAT (and not in SAT) from men with central obesity, compared to normal-weight men [36]. It was also shown that NOX is expressed to a higher extent in human VAT compared to SAT [37]. Hence, oxidative stress could be involved in the detrimental effects of fat accumulation in VAT compared to SAT, as reported in several studies [1][38][39][40][41][42].
The occurrence of oxidative stress in obesity is further demonstrated by an upregulated expression of the ROS-producing enzyme NADPH (nicotinamide adenine dinucleotide phosphate) oxidase (NOX) in AT of patients with obesity and IR [18,73,74,75]. Notably, oxidative stress in AT may be distinct for specific fat depots. For instance, higher concentrations of hydrogen peroxide, catalase and SOD activities were shown in VAT (and not in SAT) from men with central obesity, compared to normal-weight men [76]. It was also shown that NOX is expressed to a higher extent in human VAT compared to SAT [61]. Hence, oxidative stress could be involved in the detrimental effects of fat accumulation in VAT compared to SAT, as reported in several studies [10,77,78,79,80,81].
The activation of NOX is one of the principal pathways of ROS generation in AT [9][28]. NOX generates reactive species by transferring electrons from intracellular NADPH to the membrane and coupling these to molecular oxygen to produce superoxide anions, which might be further transformed into hydrogen peroxide [43][44][45]. One of the cellular targets for ROS production by NOX signaling pathway is the mitochondrion [45]. This organelle is mainly responsible for cellular energy production by oxidative phosphorylation. During oxidative phosphorylation, a small excess of electrons causes a reduction of an oxygen molecule, generating a potentially toxic free radical such as superoxide [45][46]. Given the excess nutrient availability to adipocytes in obese AT, the mitochondrial activity significantly increases, resulting in increased ROS production [47]. For instance, FFAs can promote the generation of oxygen in the mitochondrial electron transport chain by stimulating the production of reactive intermediates through protein kinase C-dependent activation of NOX [48]. Moreover, glucose overload in cells can lead to the overproduction of NADH, resulting in increased electron leakage from the mitochondrial membrane and production of superoxide [49]. Besides the excessive availability of energy substrates, higher oxidative stress in AT has been shown to induce mitochondrial dysfunction [50][51]. This exacerbates oxidative stress in AT by altering the regulation of free radical production in mitochondria and has been linked to defective fatty acid oxidation, dysregulation of adipokine secretion, and alteration of glucose homeostasis [49].Oxidative stress can therefore be both a factor sustaining the impairment of AT function and a consequence of obesity-associated metabolic dysfunctions.
The activation of NOX is one of the principal pathways of ROS generation in AT [18,68]. NOX generates reactive species by transferring electrons from intracellular NADPH to the membrane and coupling these to molecular oxygen to produce superoxide anions, which might be further transformed into hydrogen peroxide [82,83,84]. One of the cellular targets for ROS production by NOX signaling pathway is the mitochondrion [84]. This organelle is mainly responsible for cellular energy production by oxidative phosphorylation. During oxidative phosphorylation, a small excess of electrons causes a reduction of an oxygen molecule, generating a potentially toxic free radical such as superoxide [84,86]. Given the excess nutrient availability to adipocytes in obese AT, the mitochondrial activity significantly increases, resulting in increased ROS production [87]. For instance, FFAs can promote the generation of oxygen in the mitochondrial electron transport chain by stimulating the production of reactive intermediates through protein kinase C-dependent activation of NOX [88]. Moreover, glucose overload in cells can lead to the overproduction of NADH, resulting in increased electron leakage from the mitochondrial membrane and production of superoxide [89]. Besides the excessive availability of energy substrates, higher oxidative stress in AT has been shown to induce mitochondrial dysfunction [90,91]. This exacerbates oxidative stress in AT by altering the regulation of free radical production in mitochondria and has been linked to defective fatty acid oxidation, dysregulation of adipokine secretion, and alteration of glucose homeostasis [89].Oxidative stress can therefore be both a factor sustaining the impairment of AT function and a consequence of obesity-associated metabolic dysfunctions.