Cystic fibrosis (CF) is one of the most common severe autosomal recessive genetic diseases in individuals of European descent.
- cystic fibrosis
- CFTR
1. Introduction
Cystic fibrosis (CF) is one of the most common severe autosomal recessive genetic diseases in individuals of European descent. It predominantly affects the lungs and digestive system and is caused by more than 2000 mutations (www.genet.sickkids.on.ca, accessed on 1 February 2021) in the gene encoding the Cystic Fibrosis Transmembrane conductance Regulator (CFTR) protein, which regulates the hydroelectrolytic transport of exocrine, serous and mucus-secreting glands. As a consequence of these mutations, CFTR can be impaired in its functional activity, be absent or present in only a reduced quantity.
CFTR is a 168 kDa transmembrane protein (1480 amino acids) that belongs to the family of ATP-Binding Cassette transporters (ABC) [1]. These transporters share two hydrophobic transmembrane domains (TMD1 and TMD2) that constitute the pore of the channel, as well as two hydrophilic cytoplasmic ATP binding domains (NBD1 and NBD2) [2]. Among the different ABC transporters, the CFTR protein has a unique regulatory cytoplasmic domain modulated by protein kinases A and C [3]. Upon cAMP level increase, this R domain becomes phosphorylated by protein kinase A, hence allowing ATP to bind NBD1 and NBD2 domains which, through their dimerization, induce conformational changes in the transmembrane domains that result in the opening of the channel. The phenomenon is reversible since, upon ATP hydrolysis, the dissociation of NBD1-NBD2 dimers triggers the closure of the channel [4].
CFTR protein is mainly expressed at the apical membrane of epithelial cells lining the pancreas, lungs, sweat glands, intestine, liver and genital organs [5], where its major activity is to allow different anions to cross the plasma membrane, such as chloride (Cl−), bicarbonate (HCO3−) and iodide (I−) [6]. CFTR can also indirectly modulate the activity of other ionic channels, such as ENaC (Epithelial Na+ Channel) involved in the absorption of Na+ and subsequently water by epithelial cells [7]. It also plays a role in the activation and conductance of chloride channels (Outwardly Rectifying Chloride Channel, ORCC) and in reducing the activity the calcium-dependent chloride channels (Calcium-activated Chloride Channel, CaCC) [8][9][8,9]. Moreover, in the kidney epithelium, it regulates ROMK (Renal Outer Medullary Potassium) channels involved in the secretion of potassium (K+) and reabsorption of NaCl and water. Of particular interest, other studies reported that CFTR has an anti-inflammatory activity that can inhibit the transcriptional activity of NF-κB and subsequent IL-8 inflammatory cascade [10].
2. CFTR Biogenesis
CFTR is synthesized and inserted into the endoplasmic reticulum (ER) membrane in the form of a non-glycosylated precursor though CFTR mRNA translation by ER ribosomes. Folding of CFTR domains occurs independently and results in complex co-translation mechanisms and post-translational events. The first domain synthesized, which contains TM1 and TM2 segments, is TMD1. The TM2 segment is first recognized by the SRP signal recognition particle and inserted into the ER membrane. The Extra Cellular Loop 1 (ECL1) is then created by insertion of the TM1 segment. Thereafter, the other segments also work in pairs, such as TM3 with TM4 and TM5 with TM6, hence creating, respectively, the ECL2 and ECL3 peptide loops before their insertion into the ER membrane [11]. The NBD1 and R domains are then synthesized and folded followed by the TMD2 domain. As for TMD1, its insertion in the membrane occurs sequentially in pairs: segments TM7 with TM8, TM9 with TM10 and finally TM11 with TM12, hence creating ECL4, ECL5 and ECL6 loops, respectively [12]. NBD2 domain is the last one to be synthesized and folded [13][14][13,14].
Once inserted in the ER membrane, CFTR undergoes different steps of glycosylation and maturation (Figure 1) that are under the control of the Endoplasmic Reticulum Quality Control (ERQC) system where chaperone proteins like Hsp70, Hsc70, Hsp90, calnexin, Hdj-1 and Hdj-2 play major roles [15][16][17][18][15,16,17,18]. For example, the system can traffic CFTR to the Golgi by COPII vesicles [19] or trigger CFTR to the proteasome degradation pathway in cases of improper folding. This pathway depends on the Endoplasmic Reticulum-Associated Degradation (ERAD) system triggered by Hsp70 and Hsc70 [18]. From cis-, medial-, to trans-Golgi by COPI vesicles, CFTR continues to mature through post-translational modifications and specific glycosylation steps. During its maturation, three forms of CFTR are synthesized: the non-glycosylated A form (130 kDa), the core-glycosylated B form (150 kDa), which corresponds to the immature form and the fully-glycosylated C form (170 kDa), which is the mature form of the CFTR protein transported by vesicles to the plasma membrane. However, a major drawback is that CFTR biogenesis is not an efficient process with only about 25% of the synthesized proteins that are fully matured and active at the apical plasma membrane of epithelial cells. Hence, about 75% of the neosynthesized CFTR does not reach the final stage of maturation and is degraded by the ERAD coupled proteasome ubiquitin pathway (Figure 1). This peculiar property of wild-type CFTR is a direct consequence of its poor folding efficiency, a characteristic which is aggravated by mutations in its coding sequence. For example, among the many deleterious CFTR mutations, the deletion of phenylalanine at position 508 induces 99% (instead of 75% for wild-type protein) of F508del-CFTR variant to be destroyed before maturation (Figure 1) [20][21][20,21]. In 1998, a key experiment clearly demonstrated the weak folding ability of CFTR [22]. It was found that impairment of proteasome activity led to the accumulation of stable, high molecular weight, detergent-insoluble, multi-ubiquitinated forms of CFTR that were termed, for the first time, aggresomes. This term refers to an aggregation of misfolded proteins when the degradation system is overwhelmed, a prominent pathological feature also observed in neurodegenerative diseases. Additionally, a non-conventional trafficking of the CFTR protein can be activated when the conventional trafficking is blocked. As an example, ER stress has been shown to induce cell surface trafficking of the wild-type and F508-CFTR B forms via a Golgi ReAssembly Stacking Proteins (GRASP)-dependent pathway [23][24][25][23,24,25]. The detailed identification of proteins playing key roles in the processing of CFTR is of importance since they can be novel therapeutic targets [26].
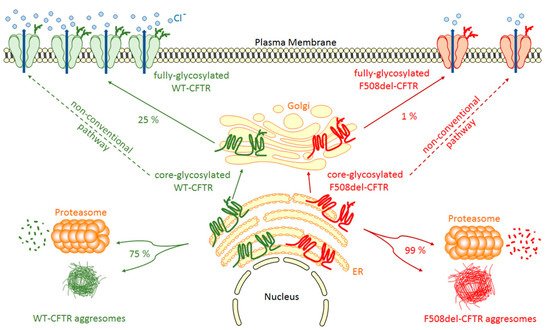
Figure 1. WT- and F508del-CFTR biogenesis. After synthesis in the Endoplasmic Reticulum (ER), the main part of core-glycosylated CFTR can form aggresomes or be recognized by the Endoplasmic Reticulum Quality Control (ERQC) system and directed to the proteasome to be degraded. CFTR escaped from ERQC undergoes different steps of glycosylation in the Golgi before being transported to the plasma membrane where it regulates ion channels. F508del-CFTR at the plasma membrane is less abundant and has only a low activity compared to WT-CFTR. A non-conventional trafficking of the CFTR protein can be activated when the conventional trafficking is blocked allowing expression of core-glycosylated CFTR at the plasma membrane.