Heme oxygenase catalyzes the rate-limiting step in heme degradation in order to generate biliverdin, carbon monoxide (CO), and iron. The inducible form of the enzyme, heme oxygenase-1 (HO-1), exerts a central role in cellular protection. The substrate, heme, is a potent pro-oxidant that can accelerate inflammatory injury and promote cell death. HO-1 has been implicated as a key mediator of inflammatory cell and tissue injury, as validated in preclinical models of acute lung injury and sepsis. A large body of work has also implicated HO-1 as a cytoprotective molecule against various forms of cell death, including necrosis, apoptosis and newly recognized regulated cell death (RCD) programs such as necroptosis, pyroptosis, and ferroptosis. While the antiapoptotic potential of HO-1 and its reaction product CO in apoptosis regulation has been extensively characterized, relatively fewer studies have explored the regulatory role of HO-1 in other forms of necrotic and inflammatory RCD (i.e., pyroptosis, necroptosis and ferroptosis). HO-1 may provide anti-inflammatory protection in necroptosis or pyroptosis. In contrast, in ferroptosis, HO-1 may play a pro-death role via enhancing iron release. HO-1 has also been implicated in co-regulation of autophagy, a cellular homeostatic program for catabolic recycling of proteins and organelles. While autophagy is primarily associated with cell survival, its occurrence can coincide with RCD programs.
- apoptosis
- autophagy
- cell death
- ferroptosis
- heme oxygenase
- necroptosis
- pyroptosis
1. Introduction
Heme oxygenase (HO-1), a vital metabolic enzyme, has emerged as a central effector of the mammalian stress response [1,2][1][2]. Early studies of microsomal metabolic activities established heme oxygenase (HO) as the rate-limiting step in heme degradation [3]. HO activity catalyzes the oxidative cleavage of heme at the α-methene bridge carbon, released as carbon monoxide (CO), to generate biliverdin-IXα (BV), while releasing the central heme iron chelate as ferrous iron (Fe II) [3]. The BV generated in the HO reaction is subsequently reduced by NAD(P)H: biliverdin reductase, to generate the lipid-soluble bile pigment bilirubin-IXα (BR) (Figure 1) [4]. Cellular HO activity is provided by two major isoforms, an inducible isozyme (HO-1) and a constitutively expressed isozyme (HO-2), which have distinct biochemical properties and arise from separate genes [5,6][5][6].
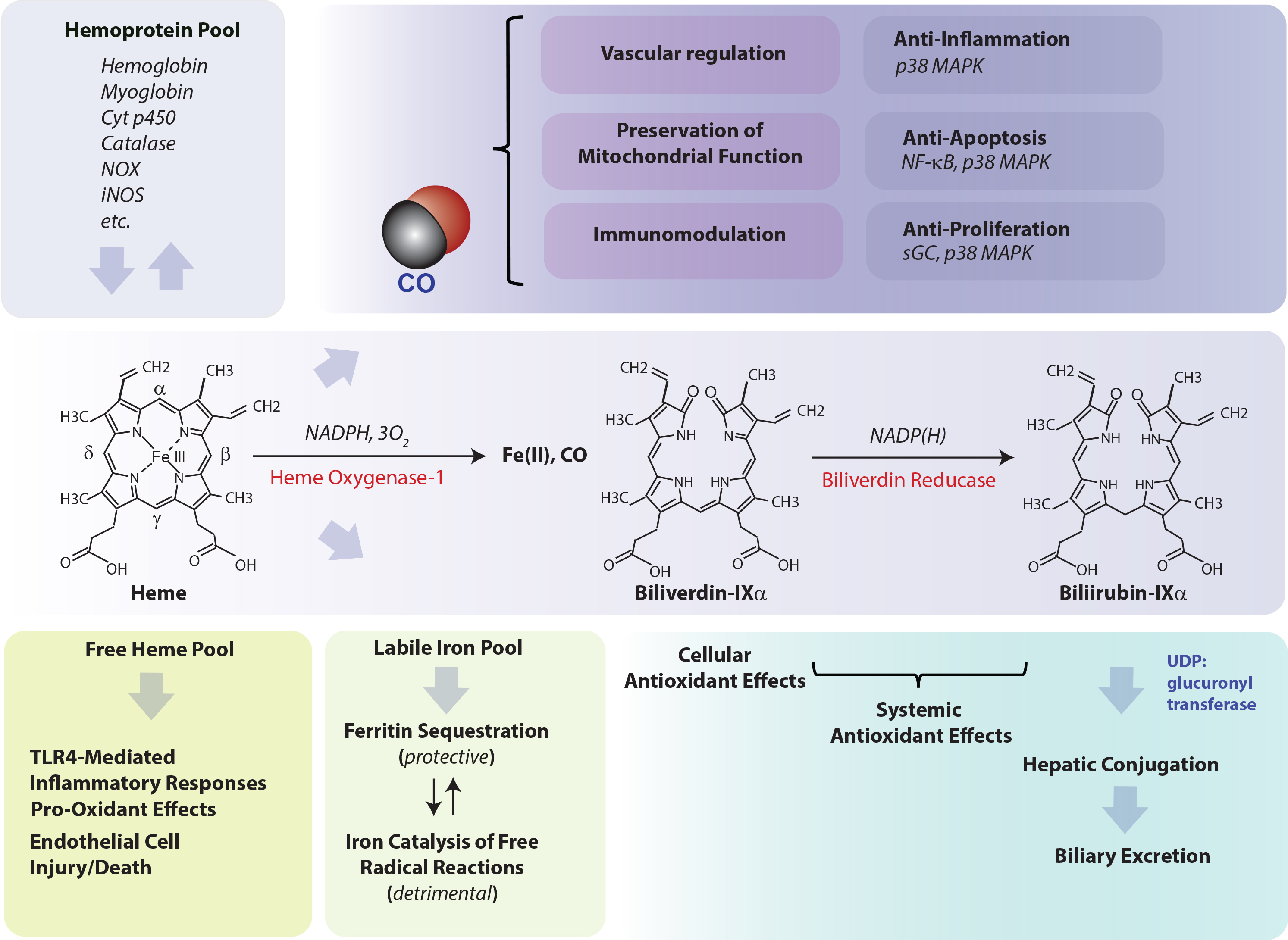
HO activity and role in cytoprotection. Heme oxygenase (HO: heme, hydrogen-donor:oxygen oxidoreductase (α-methene-oxidizing, hydroxylating), EC: 1:14:99:3) is the rate-limiting step in heme degradation. HO catalyzes the oxidative cleavage of heme at the α-methene bridge carbon, released as carbon monoxide (CO), to generate biliverdin-IXα (BV), while releasing the central heme iron chelate as ferrous iron (Fe II). Enzymatic heme degradation requires three moles of molecular oxygen (O
) and electrons derived from NADPH-cytochrome p450 reductase (EC: 1.6.2.4). The BV generated in the HO reaction is subsequently reduced by NAD(P)H: biliverdin reductase (BVR; EC: 1.3.1.24) to generate the lipid-soluble bile pigment bilirubin-IXα (BR). The source of heme for the HO reaction is derived from the turnover of hemoglobin and cellular hemoproteins. Free unbound heme released from hemolysis may represent a pro-oxidant hazard to vascular endothelium and may initiate pro-inflammatory reactions. BV and BR are known antioxidants, with circulating BR implicated as a mitigator of cardiovascular disease risk. Iron released from HO activity is equilibrated into ferritin storage, whereas unbound iron may propagate injury via catalysis of free radical-generating reactions. CO evolving from HO activity may modulate cellular function, via regulation of vascular function, immune system function, inflammation, apoptosis, and cellular proliferation. Abbreviations: CO: carbon monoxide; Cyt p450: cytochrome p450; iNOS: inducible nitric oxide synthase; NF-κB: nuclear factor-kappa-B; NOX: NADPH oxidase isoforms; p38 MAPK: p38 mitogen-activated protein kinase; sGC: soluble guanylate cyclase; TLR4: Toll-like receptor-4; UDP: uridine 5’-diphosphate.
The HO-1 field continues to attract worldwide research interest, from its mechanistic roles in regulating fundamental biological and metabolic processes, to its continuing status as a candidate therapeutic target in many disease states. Investigations in the mid-1980s established HO-1 as identical to a 32 kDa shock protein regulated by multiple forms of chemical and physical cellular stress, including oxidizing ultraviolet-A radiation, and heavy metals [7,8,9][7][8][9]. The importance of HO-1 in systemic homeostasis and iron balance was deduced from early studies using mice genetically deficient in HO-1 (Hmox1−/−). These mice were characterized by abnormal systemic iron metabolism including hepatic and renal iron deposition and anemia. Furthermore, these mice and endothelial cells derived from these mice were highly susceptible to oxidative stress [10,11][10][11]. The essential role of HO-1 in human physiology was also underscored by a unique case of HO-1 genetic deficiency in a human subject, who bore symptoms of systemic endothelial cell injury, anemia, and abnormal tissue iron accumulation [12]. Seminal studies on the macrophage inflammatory response established HO-1 as an anti-inflammatory mediator, which can limit Toll-like receptor-4 (TLR4)-dependent pro-inflammatory cytokine(s) production in activated macrophages [13]. Accumulating research since then has revealed that HO-1 can exert pleiotropic roles in mitigating inflammation, via multiple molecular mechanisms including modulation of p38 mitogen-activated protein kinase (MAPK) activity [13,14][13][14]. Furthermore, additional pioneering studies established HO-1 as an antagonist of TNF-induced endothelial cell apoptosis [15] [15].
Cell death pathways were traditionally segregated into genetically regulated and non-regulated programs (i.e., apoptosis and necrosis, respectively) [16,17,18][16][17][18]. Apoptosis requires the activation of cysteine proteases (e.g., caspases) and endonucleases without loss of plasma membrane integrity, and is morphologically characterized by cytosolic shrinkage, membrane blebbing, chromatin condensation and DNA fragmentation [19]. In contrast, necrosis was defined as an accidental or catastrophic cell death characterized by loss of energy charge, cell swelling, and plasma membrane damage resulting in the leakage of cytosolic constituents into the extracellular space, and which may trigger local inflammation and damage to surrounding tissues [18].
Further emergent discoveries have elucidated novel cell death pathways, accompanied by the paradigm-shifting revelation that certain modes of cell death that share morphological features of necrosis can also be regulated by underlying genetic programs [16]. The resulting change in cell death nomenclature now groups apoptosis with distinct forms of regulated necrosis under the term regulated cell death (RCD), to exclude non-regulated necrosis now classified as accidental cell death (ACD) [16]. The newly recognized forms of RCD include ferroptosis, pyroptosis, necroptosis, and other modalities as recently reviewed [20]. An additional genetically regulated cellular program, referred to as autophagy, represents a mechanism for cellular catabolism. Autophagy was originally classified as a cell death mode due to its coincidence with RCD and may also impact the regulation of inflammation [21].
Investigation into the role of HO-1 in cell death and related pathways was initially restricted to its largely protective role apoptosis and necrosis. Emerging studies suggest that HO-1 will have a complex modulatory or regulatory role in not only apoptosis and autophagy, but also in newly uncovered forms of RCD, namely pyroptosis, necroptosis, and/or ferroptosis.
2. Molecular Regulation of Heme Oxygenase-1
HO-1 expression responds to many diverse chemical and physical agents, including the substrate heme, a pro-oxidant compound, oxidants (e.g., H2O2), exposure to ultraviolet-A radiation, nitric oxide (NO), heavy metals and other thiol-reactive chemicals [2,7,8,9,14][2][7][8][9][14]. Further, a broad class of electrophilic plant-derived polyphenolic compounds including flavonoids and other natural antioxidants are potent inducers of HO-1 [22,23,24,25,26,27,28][22][23][24][25][26][27][28]. HO-1 responds to pro-oxidant states associated with enhanced reactive oxygen species (ROS) generation, as can be produced from dysfunctional mitochondria (mtROS) or activated inflammatory cells. Altered states of oxygen tension (pO2) above and below physiological levels can also modulate ROS production from mitochondrial metabolism. High oxygen tension (hyperoxia) increases substrate availability (O2) for enhanced mitochondrial ROS (mtROS) production, and/or increased NADPH oxidase enzymatic activity, represented by superoxide (O2−) production, and acts as a potent inducing signal for HO-1 [29]. In contrast, low pO2 (hypoxia) also favors increased ROS flux in the electron transport chain by impairing cytochrome-c oxidase activity, and selectively induces HO-1 in a species-specific manner, particularly in rodents [30,31][30][31]. HO-1 upregulation by these agents occurs mainly by transcriptional upregulation of the HMOX1 gene (Hmox1 in rodents), and results in de novo synthesis of the protein [32].
Extensive mechanistic studies have revealed that HO-1 gene regulation responds to positive regulation by nuclear factor erythroid 2-related factor-2 (Nrf2), a Cap’n’collar/basic-leucine zipper family protein that can heteromerize with small Maf proteins [33]. Nrf2 is regarded as a master regulator of the antioxidant response and regulates a series of other genes involved in detoxification. The Kelch-like ECH-associated protein (Keap1) inhibits HO-1 expression by acting as a cytoplasmic anchor for Nrf2 under basal conditions [34,35][34][35]. Keap1 enables the targeting of Nrf2 by Cullin 3-based E3 ubiquitin ligase complex, which marks Nrf2 for proteasomal degradation [36,37][36][37]. When cells are exposed to inducing stimuli, Keap1 dissociates from Nrf2, which subsequently translocates to the nucleus, where it can activate gene expression, including the Hmox1 gene [33].
Transcription factor Bach-1 acts as a transcriptional repressor of HO-1 gene expression via competition with Nrf2 [31,38,39,40][31][38][39][40]. Heme can inhibit the DNA-binding activity of Bach-1 by direct binding, as well as promote the nuclear export of Bach-1 and inhibit the proteasomal degradation of Nrf2, hereby increasing HO-1 expression [38,39,41,42][38][39][41][42]. Both Nrf2 and Bach-1 target distinct sites located in the promoter regions of Hmox1 genes. Comprehensive promoter analyses of the Hmox1 gene uncovered enhancer regions located at −4 kb and −10 kb relative to the Hmox1 transcriptional start site [43,44][43][44]. The dominant sequence element of the enhancers is the stress-responsive element (StRE), which is synonymous with the Maf response element (MARE) and antioxidant response element (ARE) [45,46][45][46]. A number of additional transcription factors have been implicated in HO-1 transcriptional regulation in a cell type-specific and inducer-specific fashion. These include AP-1 (Fos/Jun heterodimer), AP-2, heat shock factor-1 (HSF-1), hypoxia-inducible factor-1 (HIF-1), early growth-1 protein (Egr-1), nuclear factor-kappa-B (NF-κB), and cyclic AMP responsive element binding protein (CREB). The relative importance of these has been reviewed elsewhere [47,48][47][48].
3. HO-1-Mediated Cytoprotection, a Coordinated Protective Stratagem Based on Heme Removal and Heme Breakdown Product Generation
Since its discovery in 1968 [3], and its identification as a stress protein in 1988 [7], the mechanism(s) by which HO-1 can confer protection in cells and tissues, in the context of its induction by stress stimuli, remain partially understood. As the degradation of heme is the primary enzymatic function of HO-1, it stands as a valid hypothesis that its function in hemoprotein turnover and heme removal represents a cardinal mechanism underlying cytoprotection [93,94,95,96][49][50][51][52]. Indeed, heme, which has a central iron atom, has been implicated as a pro-oxidant and catalyst of free radical-generating reactions [93[49][53],97], a cytotoxic molecule with respect to vascular endothelial cells [98,99][54][55], and a pro-pathogenic mediator of diseases such as sepsis, and malaria [100,101][56][57]. Thus, the removal of heme by HO-1 may serve a context-specific protective and antioxidant function, via precluding heme from aggravating injurious or pathological processes [93,94,95][49][50][51]. By degrading heme, HO releases heme iron [1], which itself can present harmful sequelae unless detoxified, including potential catalysis of Fenton chemistry, and production of ROS and lipid peroxides [102,103][58][59]. HO-derived iron has been associated with the regulation of de novo ferritin synthesis, which in turn was associated in adaptive cytoprotection against pro-oxidant stimuli such as UVA radiation [104,105][60][61]. Ferritin is a complex multimeric molecule consisting of H and L chains which sequesters intracellular redox active iron in a crystalline core [106][62]. Ferritin has been characterized as a cytoprotective molecule in the vascular endothelium [107,108,109][63][64][65]. HO activity also releases BV which in turn is reduced to BR [3]. Both BV and BV have been shown to possess antioxidant properties in serum and bile and can attenuate free radical-generating reactions [110,111,112,113][66][67][68][69]. Much research has focused on the biological properties of CO, which originates from the α-methene bridge of heme during HO-mediated heme catalysis [3], and has emerged as an endogenous gaseous signaling mediator. CO derived from HO-1 activity was implicated in anti-inflammatory effects in macrophages based on the modulation of p38 MAPK activity. Evidence has accumulated that low concentration CO, when applied exogenously, can confer cyto- and tissue protection in inflammatory disease models in effect by influencing inflammation, apoptosis, and cell proliferation programs [2,14,114,115][2][14][70][71]. Under conditions where HO-1 is associated with cytoprotection, the pleiotropic effects of HO-1 may represent a complex cooperation of the generation and distribution of bioactive catabolic products and their downstream effects [2,14,116][2][14][72]. To achieve these cytoprotective effects, HO-1 expression must be tightly regulated. In contrast, detrimental functional roles of HO activity have been ascribed to iron overload effects [117[73][74],118], and may be relevant in neurodegenerative diseases [119][75].
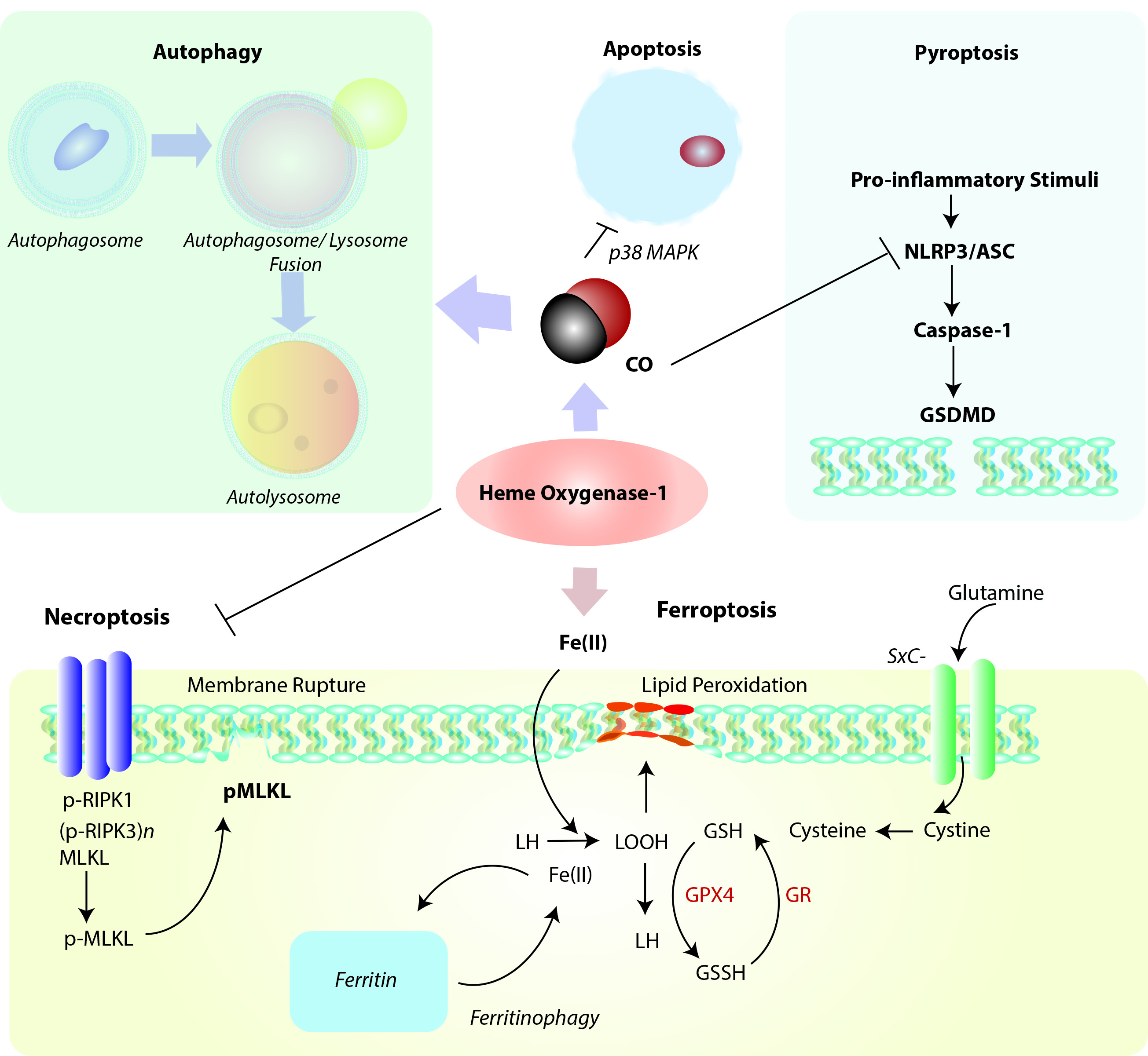
Figure 2. Positioning of HO-1 in relation to cellular homeostatic, apoptotic, and regulated cell death (RCD) programs. Heme oxygenase-1 and/or its reaction product CO can modulate the cellular autophagy program, which degrades cytosolic proteins and organelles, and is itself implicated in cellular survival/protection. HO-1 via its reaction product CO has been identified as a cellular anti-apoptotic mediator via regulation of p38 MAPK and other factors. Induction of HO-1 via either heme clearance and/or CO production may act as a mediator of inflammatory RCD programs. Among these, HO-1 derived CO may inhibit NLRP3-ASC mediated caspase-1 activation, which in term regulate gasdermin-D (GSDMD)-initiated pyroptosis. HO-1 dependent heme clearance may play a protective role in regulating RIPK3/MLKL-dependent necroptosis. Finally, HO-1 derived iron may promote lipid peroxidation, leading to ferroptotic cell death which implicates HO-1 as a pro-death regulator in the context of iron overproduction. This pathway is counter-regulated by ferritin, which sequesters iron, and promoted by autophagy-dependent degradation of ferritin (ferritinophagy). Abbreviations: ASC: apoptosis-associated speck-like protein containing caspase-activation and recruitment domain [CARD]; GSDMD: Gasdermin-D; GSH: Glutathione (reduced form); GSSG: Glutathione disulfide (oxidized form); GPX4: Glutathione peroxidase-4; GR: Glutathione reductase; LH: Lipid (reduced form), LOOH: Lipid hydroperoxide, MLKL: mixed lineage kinase domain like pseudokinase (p-, denotes phospho- form); NLRP3: nucleotide-binding domain, leucine-rich-containing family, pyrin domain-containing-3; RIPK1: Receptor-interacting serine/threonine-protein kinase 1; RIPK3: Receptor-interacting serine/threonine-protein kinase-3 (p-, denotes phospho- form); SXc-: Cystine/glutamate transporter, System Xc-.
4. HO-1 as a Regulator of Autophagy
Emerging evidence suggests that HO-1 is co-regulated with cellular autophagy (Figure 2), as both events as considered part of a global stress response. The nature of this relationship, which is supported by evidence of cross talk, remains incompletely understood and is explored in the following sections.
Cytotoxic agents that can promote oxidative stress and mitochondrial dysfunction may represent a common overlapping stimulus for both autophagy activation and HO-1 induction. Genetic studies have revealed that HO-1 can confer protection in part by regulating mitochondrial homeostasis via enhancing mitophagy and mitochondrial quality control. Cardiomyocyte-specific Hmox1 deleted mice (cm-Hmox1-/-) were highly susceptible to cardiac injury when exposed to hyperoxia challenge.173[76]. The cm-Hmox1-/- also displayed abnormal mitochondria. In hearts from these mice, both the PGC-1α and nuclear respiratory factor-1 (NRF1) signaling axis was inhibited. Further, mitochondrial biogenesis, and Pink1/Parkin-dependent mitophagy were functionally impaired in these mice.173[76] In a model of epithelial cell injury in response to cigarette smoke (CS) exposure, activation of the autophagy program correlated with epithelial cell apoptosis [77].174 In epithelial cells, viral-mediated overexpression of HO-1 reduced the expression and activation of the autophagy marker microtubule-associated protein-1 light chain 3B (LC3B), consistent with antiapoptotic cytoprotection and reduced activation of the extrinsic apoptosis pathway. siRNA-dependent HO-1 knockdown enhanced markers of autophagy and reduced cell survival in this model [77].174 These results unexpectedly demonstrated that autophagy was co-regulated with apoptosis and promoted rather than protected against cell death in response to CS exposure.
HO-1 was shown to upregulate autophagy in hepatocytes, leading to protection against hepatocyte cell death and hepatic injury from infection during sepsis in mice [78].175 HO-1 and autophagy were co-regulated in the liver in response to sepsis and inhibited hepatocyte cell death. Pharmacological inhibition of HO-1 activity or knockdown of HO-1 prevented the induction of autophagy and associated signaling in this model and resulted in increased hepatocellular injury, apoptosis, and hepatocyte death [78].175 Recent studies demonstrate that viral-mediated HO-1 overexpression can directly induce autophagy in the liver and isolated hepatocytes, and can protect against hepatotoxin exposure [79].176 Additional reports associate HO-1 dependent activation of autophagy with protection in hepatic ischemic preconditioning, I/R injury, and transplant associated I/R injury [80][81][82].177-179 HO-1 dependent autophagic signaling exerted anti-inflammatory effects in LPS-activated macrophages where HO-1 and autophagy cooperated to inhibit pro-inflammatory cytokine production [83].180 Interestingly, the p38 MAPK inhibitor SB202190 activated autophagy and induced HO-1 in endothelial cells, in a manner that could be reversed by the autophagy inhibitor Bafilomycin A1 [84].181
5. HO-1 as a modulator of Apoptosis.
Apoptosis, also formally known as Type 1 programmed cell death (PCD), is the classical genetically regulated cell death (RCD) programs initially discovered in C. Elegans. Apoptosis provides essential homeostatic functions in regulating growth and development of organs, and in tissue responses to injurious stimuli, such as exposure to xenobiotics or adverse environmental conditions [85].185 Disruption of apoptosis can promote tumorigenesis or autoimmune disease, whereas excessive apoptosis may cause organ failure. In fibroblasts, an anti-apoptotic effect was also observed with HO-1 overexpression [86].186 Application of the HO-1 reaction product CO was shown to inhibit tumor necrosis factor-α (TNFα)-initiated apoptosis in mouse fibroblasts [86],186 and endothelial cells [87].187 The antiapoptotic effect of CO in endothelial cells required the p38 MAPK pathway [87],187 and downstream activation of NF-κB [88].188 In cultured vascular smooth muscle cells, CO inhibited cytokine (TNFα, IL1-β, INFγ)-induced apoptosis, dependent on activation of soluble guanylate cyclase (sGC) [89][90].189-190 Further studies revealed similar antiapoptotic effects of CO on endothelial cell apoptosis in hyperoxia [91],122 and in anoxia/reoxygenation models [92].191 The mechanisms underlying the anti-apoptotic effects of CO have been reviewed extensively elsewhere.
6. HO-1 as a modulator of Pyroptosis.
Activation of the Nrf2/HO-1 pathway has been implicated as an inhibitor of pyroptosis in various model studies. The mechanisms by which HO-1 can mediate pyroptosis remain unclear but likely act at the level of inflammasome regulation. In model studies, exogenous CO application was found to inhibit caspase-1 activation and pro-inflammatory cytokines production in cultured macrophages in an in vitro model of NLRP3 inflammasome activation. Upregulation of HO-1 was associated with protection from LPS-induced acute kidney injury and caspase-1 dependent pyroptosis, in a mechanism involving PINK1 upregulation and preservation of mitochondrial function [93].194 In a murine model of renal I/R injury, pyroptosis was induced in injured tissue, in association with downregulation of HO-1 [94].195 Inhibition of protein arginine methylation transferase 5 (PRMT5), induced the Nrf2/HO-1 axis in conjunction with reduced tissue and cellular oxidative stress, and reduced kidney pyroptosis markers [94].195 Similarly, lung I/R injury in mice was associated with activation of pulmonary macrophage pyroptosis. The application of rHBGB1 as a preconditioning agent remediated lung injury and reduced markers of pyroptosis, in a manner dependent on activation of the Nrf2/HO-1 axis [95].196 In the hemorrhagic shock and resuscitation (HSR) model, application of CO also inhibited pyroptosis [96].197 Taken together, these studies suggest that HO-1/CO can inhibit inflammatory cell death in injury models, but more studies are needed to determine the mechanisms by which HO-1 or CO regulate pyroptosis.
7. HO-1, a mediator of necroptosis
HO-1 has been associated with cytoprotection against both apoptotic and necrotic cell death, in a dose-dependent and cell type dependent fashion. It is plausible that this protection would extend to regulated forms of necrosis (necroptosis). To date however, only few studies have examined the relationship between HO-1 and necroptosis in cellular and injury models. Free heme is a cytotoxic agent, which can act as a pro-oxidant, via its central iron chelate. Heme released from hemoglobin can injury endothelial cells of the vasculature by causing membrane damage. Pro-inflammatory effects of heme have been implicated in the pathogenesis of sepsis and malaria. In vitro experiments showed that heme treatment of macrophages can cause macrophage cell death with morphological features of necrosis. The authors found that heme-induced necrotic cell death was dependent on TLR4 regulated TNF production and enhanced ROS generation. Applications of antioxidants and JNK inhibitors or of necrostatin-1, a selective inhibitor of receptor-interacting protein 1 (RIPK1) was protective. Similarly, cells genetically deficient in Ripk1 or Ripk3 were protected from heme-induced cell death. Macrophages from Hmox1−/− mice were also more sensitive to heme toxicity and oxidative stress [97].207
8. HO-1, a mediator of ferroptosis
Ferroptosis is defined as a uniquely iron-dependent necrotic form of necrotic cell death that is distinct from autophagy, apoptosis and other forms of necrosis-like RCD, including necroptosis, and which has been implicated in the propagation of inflammation [98][99].211-212 The morphologically distinct features of ferroptosis include mitochondrial shrinkage and increased mitochondrial membrane density [98].211 Blockage of cystine uptake is a primary stimulator of ferroptosis, which results in impaired synthesis of reduced glutathione (GSH) for use as substrate for (phospholipid-hydroperoxide) glutathione peroxidase-4 (GPX4) mediated detoxification of organic hydroperoxides [100].213 Iron can catalyze the peroxidation of lipids resulting in membrane disruption characteristic of ferroptosis. Inhibition of cystine uptake also promotes the degradation of ferritin via a nuclear receptor coactivator 4 (NCOA4)-mediated selective autophagy mechanism [101][102].214-215 Ferroptotic cell death is inhibited by lipophilic antioxidants, such as ferrostatin-1 and others [103];216 and by iron chelators [104],217 which also can contextually inhibit oxidant stimulated HO-1 expression [105][106].218-219 Although HO-1 is generally found to be protective in autophagy and other types of RCD, its specific role in ferroptosis remains unclear. While HO-1 may be protective in mitigating pro-oxidant states by preserving mitochondrial function, as implicated in the initiation of inflammatory cell death and apoptosis, HO-1 releases iron as a reaction by-product, which is thereby implicated in the initiation of ferroptosis if in excess or left unsequestered by ferritin. Thus, whether HO-1 is regarded as a promoter or inhibitor of ferroptosis is context-dependent and varies with model studies.
References
- Abraham, N.G.; Kappas, A. Pharmacological and clinical aspects of heme oxygenase. Pharmacol. Rev. 2008, 60, 79–127.
- Ryter, S.W.; Alam, J.; Choi, A.M. Heme oxygenase-1/carbon monoxide: From basic science to therapeutic applications. Physiol. Rev. 2006, 86, 583–650.
- Tenhunen, R.; Marver, H.S.; Schmid, R. Microsomal heme oxygenase. Characterization of the enzyme. J. Biol. Chem. 1969, 244, 6388–6394.
- Tenhunen, R.; Ross, M.E.; Marver, H.S.; Schmid, R. Reduced nicotinamide-adenine dinucleotide phosphate dependent biliverdin reductase: Partial purification and characterization. Biochemistry 1970, 9, 298–303.
- Maines, M.D. The heme oxygenase system: A regulator of second messenger gases. Annu. Rev. Pharmacol. Toxicol. 1997, 37, 517–554.
- Cruse, I.; Maines, M.D. Evidence suggesting that the two forms of heme oxygenase are products of different genes. J. Biol. Chem. 1988, 263, 3348–3353.
- Keyse, S.M.; Tyrrell, R.M. Heme oxygenase is the major 32-kDa stress protein induced in human skin fibroblasts by UVA radiation, hydrogen peroxide, and sodium arsenite. Proc. Natl. Acad. Sci. USA 1989, 86, 99–103.
- Shibahara, S.; Müller, R.; Taguchi, H.; Yoshida, T. Cloning and expression of cDNA for rat heme oxygenase. Proc. Natl. Acad. Sci. USA 1985, 82, 7865–7869.
- Alam, J.; Shibahara, S.; Smith, A. Transcriptional activation of the heme oxygenase gene by heme and cadmium in mouse hepatoma cells. J. Biol. Chem. 1989, 264, 6371–6375.
- Poss, K.D.; Tonegawa, S. Heme oxygenase-1 is required for mammalian iron reutilization. Proc. Natl. Acad. Sci. USA 1997, 94, 10919–10924.
- Poss, K.D.; Tonegawa, S. Reduced stress defense in heme oxygenase 1-deficient cells. Proc. Natl. Acad. Sci. USA 1997, 94, 10925–10930.
- Yachie, A.; Niida, Y.; Wada, T.; Igarashi, N.; Kaneda, H.; Toma, T.; Ohta, K.; Kasahara, Y.; Koizumi, S. Oxidative stress causes enhanced endothelial cell injury in human heme oxygenase-1 deficiency. J Clin. Invest. 1999, 103, 129–135.
- Otterbein, L.E.; Bach, F.H.; Alam, J.; Soares, M.; Tao Lu, H.; Wysk, M.; Davis, R.J.; Flavell, R.A.; Choi, A.M. Carbon monoxide has anti-inflammatory effects involving the mitogen-activated protein kinase pathway. Nat. Med. 2000, 6, 422–428.
- Ryter, S.W.; Choi, A.M.K. Targeting heme oxygenase-1 and carbon monoxide for therapeutic modulation of inflammation. Transl. Res. 2016, 167, 7–34.
- Brouard, S.; Otterbein, L.E.; Anrather, J.; Tobiasch, E.; Bach, F.H.; Choi, A.M.; Soares, M.P. Carbon monoxide generated by heme oxygenase-1 suppresses endothelial cell apoptosis. J. Exp. Med. 2000, 192, 1015–1026.
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541.
- Galluzzi, L.; Maiuri, M.C.; Vitale, I.; Zischka, H.; Castedo, M.; Zitvogel, L.; Kroemer, G. Cell death modalities: Classification and pathophysiological implications. Cell Death Differ. 2007, 14, 1237–1243.
- Majno, G.; Joris, I. Apoptosis, oncosis, and necrosis. An overview of cell death. Am. J Pathol. 1995, 146, 3–15.
- Kroemer, G.; Dallaporta, B.; Resche-Rigon, M. The mitochondrial death/life regulator in apoptosis and necrosis. Annu. Rev. Physiol. 1998, 60, 619–642.
- Kist, M.; Vucic, D. Cell death pathways: Intricate connections and disease implications. EMBO J. 2021, e106700.
- Galluzzi, L.; Baehrecke, E.H.; Ballabio, A.; Boya, P.; Bravo-San Pedro, J.M.; Cecconi, F.; Choi, A.M.; Chu, C.T.; Codogno, P.; Colombo, M.I.; et al. Molecular definitions of autophagy and related processes. EMBO J. 2017, 36, 1811–1836.
- Pittala, V.; Vanella, L.; Salerno, L.; Romeo, G.; Marrazzo, A.; Di Giacomo, C.; Sorrenti, V. Effects of polyphenolic derivatives on heme oxygenase-system in metabolic dysfunctions. Curr. Med. Chem. 2018, 25, 1577–1595.
- Hahn, D.; Shin, S.H.; Bae, J.S. Natural antioxidant and anti-inflammatory compounds in foodstuff or medicinal herbs inducing heme oxygenase-1 expression. Antioxidants 2020, 9, 1191.
- Bajpai, V.K.; Alam, M.B.; Ju, M.K.; Kwon, K.R.; Huh, Y.S.; Han, Y.K.; Lee, S.H. Antioxidant mechanism of polyphenol-rich Nymphaea nouchali leaf extract protecting DNA damage and attenuating oxidative stress-induced cell death via Nrf2-mediated heme-oxygenase-1 induction coupled with ERK/p38 signaling pathway. Biomed. Pharmacother. 2018, 103, 1397–1407.
- Scapagnini, G.; Foresti, R.; Calabrese, V.; Giuffrida Stella, A.M.; Green, C.J.; Motterlini, R. Caffeic acid phenethyl ester and curcumin: A novel class of heme oxygenase-1 inducers. Mol. Pharmacol. 2002, 61, 554–556.
- Martin, D.; Rojo, A.I.; Salinas, M.; Diaz, R.; Gallardo, G.; Alam, J.; De Galarreta, C.M.; Cuadrado, A. Regulation of heme oxygenase-1 expression through the phosphatidylinositol 3-kinase/Akt pathway and the Nrf2 transcription factor in response to the antioxidant phytochemical carnosol. J. Biol. Chem. 2004, 279, 8919–8929.
- Foresti, R.; Hoque, M.; Monti, D.; Green, C.J.; Motterlini, R. Differential activation of heme oxygenase-1 by chalcones and rosolic acid in endothelial cells. J. Pharmacol. Exp. Ther. 2005, 312, 686–693.
- Ogborne, R.M.; Rushworth, S.A.; Charalambos, C.; O’Connell, M.A. Haem oxygenase-1: A target for dietary antioxidants. Biochem. Soc. Trans. 2004, 32, 1003–1005.
- Lee, P.J.; Alam, J.; Sylvester, S.L.; Inamdar, N.; Otterbein, L.; Choi, A.M. Regulation of heme oxygenase-1 expression in vivo and in vitro in hyperoxic lung injury. Am. J. Respir. Cell Mol. Biol. 1996, 14, 556–568.
- Lee, P.J.; Jiang, B.H.; Chin, B.Y.; Iyer, N.V.; Alam, J.; Semenza, G.L.; Choi, A.M. Hypoxia-inducible factor-1 mediates transcriptional activation of the heme oxygenase-1 gene in response to hypoxia. J. Biol. Chem. 1997, 272, 5375–5381.
- Kitamuro, T.; Takahashi, K.; Ogawa, K.; Udono-Fujimori, R.; Takeda, K.; Furuyama, K.; Nakayama, M.; Sun, J.; Fujita, H.; Hida, W.; et al. Bach1 functions as a hypoxia-inducible repressor for the heme oxygenase-1 gene in human cells. J. Biol. Chem. 2003, 278, 9125–9133.
- Keyse, S.M.; Applegate, L.A.; Tromvoukis, Y.; Tyrrell, R.M. Oxidant stress leads to transcriptional activation of the human heme oxygenase gene in cultured skin fibroblasts. Mol. Cell Biol. 1990, 10, 4967–4969.
- Alam, J.; Stewart, D.; Touchard, C.; Boinapally, S.; Choi, A.M.; Cook, J.L. Nrf2, a Cap’n’Collar transcription factor, regulates induction of the heme oxygenase-1 gene. J. Biol. Chem. 1999, 274, 26071–26078.
- Kang, M.I.; Kobayashi, A.; Wakabayashi, N.; Kim, S.G.; Yamamoto, M. Scaffolding of Keap1 to the actin cytoskeleton controls the function of Nrf2 as key regulator of cytoprotective phase 2 genes. Proc. Natl. Acad. Sci. USA 2004, 101, 2046–2051.
- Zipper, L.M.; Mulcahy, R.T. The Keap1 BTB/POZ dimerization function is required to sequester Nrf2 in cytoplasm. J. Biol. Chem. 2002, 277, 36544–36552.
- Itoh, K.; Wakabayashi, N.; Katoh, Y.; Ishii, T.; O’Connor, T.; Yamamoto, M. Keap1 regulates both cytoplasmic-nuclear shuttling and degradation of Nrf2 in response to electrophiles. Genes Cells 2003, 8, 379–391.
- Itoh, K.; Wakabayashi, N.; Katoh, Y.; Ishii, T.; Igarashi, K.; Engel, J.D.; Yamamoto, M. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev. 1999, 13, 76–86.
- Igarashi, K.; Sun, J. The heme-Bach1 pathway in the regulation of oxidative stress response and erythroid differentiation. Antioxid. Redox Signal. 2006, 8, 107–118.
- Sun, J.; Hoshino, H.; Takaku, K.; Nakajima, O.; Muto, A.; Suzuki, H.; Tashiro, S.; Takahashi, S.; Shibahara, S.; Alam, J.; et al. Hemoprotein Bach1 regulates enhancer availability of heme oxygenase-1 gene. EMBO J. 2002, 21, 5216–5224.
- Oyake, T.; Itoh, K.; Motohashi, H.; Hayashi, N.; Hoshino, H.; Nishizawa, M.; Yamamoto, M.; Igarashi, K. Bach proteins belong to a novel family of BTB-basic leucine zipper transcription factors that interact with MafK and regulate transcription through the NF-E2 site. Mol. Cell Biol. 1996, 16, 6083–6095.
- Sun, J.; Brand, M.; Zenke, Y.; Tashiro, S.; Groudine, M.; Igarashi, K. Heme regulates the dynamic exchange of Bach1 and NF-E2-related factors in the Maf transcription factor network. Proc. Natl. Acad. Sci. USA 2004, 101, 1461–1466.
- Alam, J.; Killeen, E.; Gong, P.; Naquin, R.; Hu, B.; Stewart, D.; Ingelfinger, J.R.; Nath, K.A. Heme activates the heme oxygenase-1 gene in renal epithelial cells by stabilizing Nrf2. Am. J. Physiol. Renal Physiol. 2003, 284, F743–F752.
- Alam, J.; Cai, J.; Smith, A. Isolation and characterization of the mouse heme oxygenase-1 gene. Distal 5’ sequences are required for induction by heme or heavy metals. J. Biol. Chem. 1994, 269, 1001–1009.
- Alam, J.; Camhi, S.; Choi, A.M. Identification of a second region upstream of the mouse heme oxygenase-1 gene that functions as a basal level and inducer-dependent transcription enhancer. J. Biol. Chem. 1995, 270, 11977–11984.
- Inamdar, N.M.; Ahn, Y.I.; Alam, J. The heme-responsive element of the mouse heme oxygenase-1 gene is an extended AP-1 binding site that resembles the recognition sequences for MAF and NF-E2 transcription factors. Biochem. Biophys. Res. Commun. 1996, 221, 570–576.
- Alam, J.; Cook, J.L. How many transcription factors does it take to turn on the heme oxygenase-1 gene? Am. J. Respir. Cell Mol. Biol. 2007, 36, 166–174.
- Alam, J.; Igarashi, K.; Immenschuh, S.; Shibahara, S.; Tyrrell, R.M. Regulation of heme oxygenase-1 gene transcription: Recent advances and highlights from the International Conference (Uppsala, 2003) on Heme Oxygenase. Antioxid. Redox Signal. 2004, 6, 924–933.
- Morse, D.; Lin, L.; Choi, A.M.; Ryter, S.W. Heme oxygenase-1, a critical arbitrator of cell death pathways in lung injury and disease. Free Radic. Biol. Med. 2009, 47, 1–12.
- Haines, D.D.; Tosaki, A. Heme degradation in pathophysiology of and countermeasures to inflammation-associated disease. Int. J. Mol. Sci. 2020, 21, 9698.
- Ryter, S.W.; Tyrrell, R.M. The heme synthesis and degradation pathways: Role in oxidant sensitivity. Heme oxygenase has both pro- and antioxidant properties. Free Radic. Biol. Med. 2000, 28, 289–309.
- Immenschuh, S.; Vijayan, V.; Janciauskiene, S.; Gueler, F. Heme as a target for therapeutic interventions. Front. Pharmacol. 2017, 8, 146.
- Donegan, R.K.; Moore, C.M.; Hanna, D.A.; Reddi, A.R. Handling heme: The mechanisms underlying the movement of heme within and between cells. Free Radic. Biol. Med. 2019, 133, 88–100.
- Kumar, A.; Ganini, D.; Deterding, L.J.; Ehrenshaft, M.; Chatterjee, S.; Mason, R.P. Immuno-spin trapping of heme-induced protein radicals: Implications for heme oxygenase-1 induction and heme degradation. Free Radic. Biol. Med. 2013, 61, 265–272.
- Jeney, V.; Balla, J.; Yachie, A.; Varga, Z.; Vercellotti, G.M.; Eaton, J.W.; Balla, G. Pro-oxidant and cytotoxic effects of circulating heme. Blood 2002, 100, 879–887.
- Balla, J.; Jacob, H.S.; Balla, G.; Nath, K.; Eaton, J.W.; Vercellotti, G.M. Endothelial-cell heme uptake from heme proteins: Induction of sensitization and desensitization to oxidant damage. Proc. Natl. Acad. Sci. USA 1993, 90, 9285–9289.
- Larsen, R.; Gozzelino, R.; Jeney, V.; Tokaji, L.; Bozza, F.A.; Japiassú, A.M.; Bonaparte, D.; Cavalcante, M.M.; Chora, A.; Ferreira, A.; et al. A central role for free heme in the pathogenesis of severe sepsis. Sci. Transl. Med. 2010, 2, 51ra71.
- Pereira, M.L.M.; Marinho, C.R.F.; Epiphanio, S. Could heme oxygenase-1 be a new target for therapeutic intervention in malaria-associated acute lung injury/acute respiratory distress syndrome? Front. Cell Infect. Microbiol. 2018, 8, 161.
- Halliwell, B.; Gutteridge, J.M. Oxygen toxicity, oxygen radicals, transition metals and disease. Biochem. J. 1984, 219, 1–14.
- Dröge, W. Free radicals in the physiological control of cell function. Physiol. Rev. 2002, 82, 47–95.
- Vile, G.F.; Tyrrell, R.M. Oxidative stress resulting from ultraviolet A irradiation of human skin fibroblasts leads to a heme oxygenase-dependent increase in ferritin. J. Biol. Chem. 1993, 268, 14678–14681.
- Vile, G.F.; Basu-Modak, S.; Waltner, C.; Tyrrell, R.M. Heme oxygenase 1 mediates an adaptive response to oxidative stress in human skin fibroblasts. Proc. Natl. Acad. Sci. USA 1994, 91, 2607–2610.
- Arosio, P.; Levi, S. Ferritin, iron homeostasis, and oxidative damage. Free Radic. Biol. Med. 2002, 33, 457–463.
- Balla, G.; Jacob, H.S.; Balla, J.; Rosenberg, M.; Nath, K.; Apple, F.; Eaton, J.W.; Vercellotti, G.M. Ferritin: A cytoprotective antioxidant strategem of endothelium. J. Biol. Chem. 1992, 267, 18148–18153.
- Juckett, M.B.; Balla, J.; Balla, G.; Jessurun, J.; Jacob, H.S.; Vercellotti, G.M. Ferritin protects endothelial cells from oxidized low density lipoprotein in vitro. Am. J. Pathol. 1995, 147, 782–789.
- Balla, J.; Nath, K.A.; Balla, G.; Juckett, M.B.; Jacob, H.S.; Vercellotti, G.M. Endothelial cell heme oxygenase and ferritin induction in rat lung by hemoglobin in vivo. Am. J. Physiol. 1995, 268, L321–L327.
- Stocker, R.; Yamamoto, Y.; McDonagh, A.F.; Glazer, A.N.; Ames, B.N. Bilirubin is an antioxidant of possible physiological importance. Science 1987, 235, 1043–1046.
- Neuzil, J.; Stocker, R. Bilirubin attenuates radical-mediated damage to serum albumin. FEBS Lett. 1993, 331, 281–284.
- Stocker, R.; Glazer, A.N.; Ames, B.N. Antioxidant activity of albumin-bound bilirubin. Proc. Natl. Acad. Sci USA 1987, 84, 5918–5922.
- Stocker, R.; Ames, B.N. Potential role of conjugated bilirubin and copper in the metabolism of lipid peroxides in bile. Proc. Natl. Acad. Sci USA 1987, 84, 8130–8134.
- Motterlini, R.; Otterbein, L.E. The therapeutic potential of carbon monoxide. Nat. Rev. Drug Discov. 2010, 9, 728–743.
- Ryter, S.W.; Ma, K.C.; Choi, A.M.K. Carbon monoxide in lung cell physiology and disease. Am. J. Physiol. Cell Physiol. 2018, 314, C211–C227.
- Otterbein, L.E.; Choi, A.M. Heme oxygenase: Colors of defense against cellular stress. Am. J. Physiol. Lung Cell. Mol. Physiol. 2000, 279, L1029–L1037.
- Kvam, E.; Hejmadi, V.; Ryter, S.; Pourzand, C.; Tyrrell, R.M. Heme oxygenase activity causes transient hypersensitivity to oxidative ultraviolet A radiation that depends on release of iron from heme. Free Radic. Biol. Med. 2000, 28, 1191–1196.
- Suttner, D.M.; Dennery, P.A. Reversal of HO-1 related cytoprotection with increased expression is due to reactive iron. FASEB J. 1999, 13, 1800–1809.
- Schipper, H.M.; Song, W.; Tavitian, A.; Cressatti, M. The sinister face of heme oxygenase-1 in brain aging and disease. Prog. Neurobiol. 2019, 172, 40–70.
- Peng, Z.; Liao, Y.; Wang, X.; Chen, L.; Wang, L.; Qin, C.; Wang, Z.; Cai, M.; Hu, J.; Li, D.; et al. Heme oxygenase-1 regulates autophagy through carbon-oxygen to alleviate deoxynivalenol-induced hepatic damage. Arch. Toxicol. 2020, 94, 573–588.
- Liu, A.; Guo, E.; Yang, J.; Li, R.; Yang, Y.; Liu, S.; Hu, J.; Jiang, X.; Dirsch, O.; Dahmen, U.; et al. Ischemic preconditioning attenuates ischemia/reperfusion injury in rat steatotic liver: Role of heme oxygenase-1-mediated autophagy. Oncotarget 2016, 7, 78372–78386.
- Yun, N.; Cho, H.I.; Lee, S.M. Impaired autophagy contributes to hepatocellular damage during ischemia/reperfusion: Heme oxygenase-1 as a possible regulator. Free Radic. Biol. Med. 2014, 68, 168–177.
- Nakamura, K.; Kageyama, S.; Yue, S.; Huang, J.; Fujii, T.; Ke, B.; Sosa, R.A.; Reed, E.F.; Datta, N.; Zarrinpar, A.; et al. Heme oxygenase-1 regulates sirtuin-1-autophagy pathway in liver transplantation: From mouse to human. Am. J. Transplant. 2018, 18, 1110–1121.
- Waltz, P.; Carchman, E.H.; Young, A.C.; Rao, J.; Rosengart, M.R.; Kaczorowski, D.; Zuckerbraun, B.S. Lipopolysaccaride induces autophagic signaling in macrophages via a TLR4, heme oxygenase-1 dependent pathway. Autophagy 2011, 7, 315–320.
- Schwartz, M.; Böckmann, S.; Borchert, P.; Hinz, B. SB202190 inhibits endothelial cell apoptosis via induction of autophagy and heme oxygenase-1. Oncotarget 2018, 9, 23149–23163.
- Lee, S.J.; Ryter, S.W.; Xu, J.F.; Nakahira, K.; Kim, H.P.; Choi, A.M.; Kim, Y.S. Carbon monoxide activates autophagy via mitochondrial reactive oxygen species formation. Am. J. Respir. Cell Mol. Biol. 2011, 45, 867–873.
- Lee, S.; Lee, S.J.; Coronata, A.A.; Fredenburgh, L.E.; Chung, S.W.; Perrella, M.A.; Nakahira, K.; Ryter, S.W.; Choi, A.M. Carbon monoxide confers protection in sepsis by enhancing beclin 1-dependent autophagy and phagocytosis. Antioxid. Redox Signal. 2014, 20, 432–442.
- Chen, S.; Ding, R.; Hu, Z.; Yin, X.; Xiao, F.; Zhang, W.; Yan, S.; Lv, C. MicroRNA-34a inhibition alleviates lung injury in cecal ligation and puncture Induced septic mice. Front. Immunol. 2020, 11, 1829.
- Liu, X.M.; Chapman, G.B.; Peyton, K.J.; Schafer, A.I.; Durante, W. Carbon monoxide inhibits apoptosis in vascular smooth muscle cells. Cardiovasc. Res. 2002, 55, 396–405.
- Zhang, X.; Shan, P.; Otterbein, L.E.; Alam, J.; Flavell, R.A.; Davis, R.J.; Choi, A.M.; Lee, P.J. Carbon monoxide inhibition of apoptosis during ischemia-reperfusion lung injury is dependent on the p38 mitogen-activated protein kinase pathway and involves caspase 3. J. Biol. Chem. 2003, 278, 1248–1258.
- Lamkanfi, M.; Dixit, V.M. Mechanisms and functions of inflammasomes. Cell 2014, 157, 1013–1022.
- Jorgensen, I.; Miao, E.A. Pyroptotic cell death defends against intracellular pathogens. Immunol. Rev. 2015, 265, 130–142.
- Li, H.B.; Zhang, X.Z.; Sun, Y.; Zhou, Q.; Song, J.N.; Hu, Z.F.; Li, Y.; Wu, J.N.; Guo, Y.; Zhang, Y.; et al. HO-1/PINK1 Regulated mitochondrial fusion/fission to inhibit pyroptosis and attenuate septic acute kidney injury. Biomed. Res. Int. 2020, 2020, 2148706.
- Diao, C.; Chen, Z.; Qiu, T.; Liu, H.; Yang, Y.; Liu, X.; Wu, J.; Wang, L. Inhibition of PRMT5 attenuates oxidative stress-induced pyroptosis via activation of the Nrf2/HO-1 signal pathway in a mouse model of Renal Ischemia-Reperfusion Injury. Oxid. Med. Cell Longev. 2019, 2019, 2345658.
- Wang, X.; Wang, Y.; Kim, H.P.; Nakahira, K.; Ryter, S.W.; Choi, A.M. Carbon monoxide protects against hyperoxia-induced endothelial cell apoptosis by inhibiting reactive oxygen species formation. J. Biol. Chem. 2007, 282, 1718–1726.
- Fei, L.; Jingyuan, X.; Fangte, L.; Huijun, D.; Liu, Y.; Ren, J.; Jinyuan, L.; Linghui, P. Preconditioning with rHMGB1 ameliorates lung ischemia-reperfusion injury by inhibiting alveolar macrophage pyroptosis via the Keap1/Nrf2/HO-1 signaling pathway. J. Transl. Med. 2020, 18, 301.
- Choi, M.E.; Price, D.R.; Ryter, S.W.; Choi, A.M.K. Necroptosis: A crucial pathogenic mediator of human disease. JCI Insight. 2019, 4, e128834.
- Linkermann, A.; Green, D.R. Necroptosis. N. Engl. J. Med. 2014, 370, 455–465.
- Kaczmarek, A.; Vandenabeele, P.; Krysko, D.V. Necroptosis: The release of damage-associated molecular patterns and its physiological relevance. Immunity. 2013, 38, 209–223.
- Tonnus, W.; Meyer, C.; Paliege, A.; Belavgeni, A.; von Mässenhausen, A.; Bornstein, S.R.; Hugo, C.; Becker, J.U.; Linkermann, A. The pathological features of regulated necrosis. J. Pathol. 2019, 247, 697–707.
- Sun, Y.; Chen, P.; Zhai, B.; Zhang, M.; Xiang, Y.; Fang, J.; Xu, S.; Gao, Y.; Chen, X.; Sui, X.; et al. The emerging role of ferroptosis in inflammation. Biomed. Pharmacother. 2020, 127, 110108.
- Kajarabille, N.; Latunde-Dada, G.O. Programmed cell-death by ferroptosis: Antioxidants as mitigators. Int. J. Mol. Sci. 2019, 20, 4968.
- Yu, H.; Guo, P.; Xie, X.; Wang, Y.; Chen, G. Ferroptosis, a new form of cell death, and its relationships with tumourous diseases. J. Cell. Mol. Med. 2017, 21, 648–657.
- Ryter, S.W.; Si, M.; Lai, C.C.; Su, C.Y. Regulation of endothelial heme oxygenase activity during hypoxia is dependent on chelatable iron. Am. J. Physiol. Heart Circ. Physiol. 2000, 279, H2889–H2897.
- Keyse, S.M.; Tyrrell, R.M. Induction of the heme oxygenase gene in human skin fibroblasts by hydrogen peroxide and UVA (365 nm) radiation: Evidence for the involvement of the hydroxyl radical. Carcinogenesis 1990, 11, 787–791.
- Fang, X.; Wang, H.; Han, D.; Xie, E.; Yang, X.; Wei, J.; Gu, S.; Gao, F.; Zhu, N.; Yin, X.; et al. Ferroptosis as a target for protection against cardiomyopathy. Proc. Natl. Acad. Sci. USA 2019, 116, 2672–2680.
- Bao, W.D.; Zhou, X.T.; Zhou, L.T.; Wang, F.; Yin, X.; Lu, Y.; Zhu, L.Q.; Liu, D. Targeting miR-124/Ferroportin signaling ameliorated neuronal cell death through inhibiting apoptosis and ferroptosis in aged intracerebral hemorrhage murine model. Aging Cell 2020, 19, e13235.
- Kwon, M.Y.; Park, E.; Lee, S.J.; Chung, S.W. Heme oxygenase-1 accelerates erastin-induced ferroptotic cell death. Oncotarget 2015, 6, 24393–24403.
- Chang, L.C.; Chiang, S.K.; Chen, S.E.; Yu, Y.L.; Chou, R.H.; Chang, W.C. Heme oxygenase-1 mediates BAY 11-7085 induced ferroptosis. Cancer Lett. 2018, 416, 124–137.
- Guerrero-Hue, M.; García-Caballero, C.; Palomino-Antolín, A.; Rubio-Navarro, A.; Vázquez-Carballo, C.; Herencia, C.; Martín-Sanchez, D.; Farré-Alins, V.; Egea, J.; Cannata, P.; et al. Curcumin reduces renal damage associated with rhabdomyolysis by decreasing ferroptosis-mediated cell death. FASEB J. 2019, 33, 8961–8975.