Complement 5 (C5) protein, the cleavage of which is mediated by the serine proteases C5 convertases to generate two different fragments: C5a, which is a potent chemoattractant and pro-inflammatory modulator, and C5b, which initiates the formation of MAC, mediating cell lysis and triggering inflammation.
Complement 5 (C5) protein, the cleavage of which is mediated by the serine proteases C5 convertases to generate two different fragments [3]: C5a, which is a potent chemoattractant and pro-inflammatory modulator [4,5], and C5b, which initiates the formation of MAC, mediating cell lysis and triggering inflammation.
- complement system
- C5a/C5aR axis
- C5a receptor1
- neuropathies
- C5a inhibitor
1. Introduction
The complement system is a crucial element of the innate immune response that works in concert with antibodies and phagocytic cells to clear pathogens [1]. It consists of a number of precursor proteins that are cleaved by specific proteases to generate various complement peptides and fragments, ultimately leading to the formation of the Membrane Attack Complex (MAC) [2]. One of the key components of the complement system is the Complement 5 (C5) protein, the cleavage of which is mediated by the serine proteases C5 convertases to generate two different fragments [3]: C5a, which is a potent chemoattractant and pro-inflammatory modulator [4][5], and C5b, which initiates the formation of MAC, mediating cell lysis and triggering inflammation [6]. When properly activated, C5a is crucial for host defence system and clearance of pathogens; however, its inappropriate activation is involved in a wide range of disorders, including peripheral neuropathic diseases [7][8].
The complement system is a crucial element of the innate immune response that works in concert with antibodies and phagocytic cells to clear pathogens [1]. It consists of a number of precursor proteins that are cleaved by specific proteases to generate various complement peptides and fragments, ultimately leading to the formation of the Membrane Attack Complex (MAC) [2]. One of the key components of the complement system is the Complement 5 (C5) protein, the cleavage of which is mediated by the serine proteases C5 convertases to generate two different fragments [3]: C5a, which is a potent chemoattractant and pro-inflammatory modulator [4,5], and C5b, which initiates the formation of MAC, mediating cell lysis and triggering inflammation [6]. When properly activated, C5a is crucial for host defence system and clearance of pathogens; however, its inappropriate activation is involved in a wide range of disorders, including peripheral neuropathic diseases [7,8].
2. Complement Pathways
The complement is a major component of the innate immune system and acts as a bridge between innate and acquired immunity. Over the years, it has become clear that the complement has various functions, ranging from the mediation of inflammatory responses to the regulation of host cell clearance after their programmed cell death [9], and takes part in nearly every step of the immune reaction. It is composed of over 50 proteins [10]. Among these, the soluble ones are produced mainly by the liver and can be detected in the plasma and on cell surfaces as inactive precursors (zymogens) [1]; their cleavage by serine proteases activates a cascade of enzymatic reactions that is tightly regulated to assure complement activation is triggered only at specific locations, thus avoiding host tissue damage. Activation of the complement system occurs through three distinct pathways: the classical (CP), lectin (LP), and alternative (AP) pathways [2] ().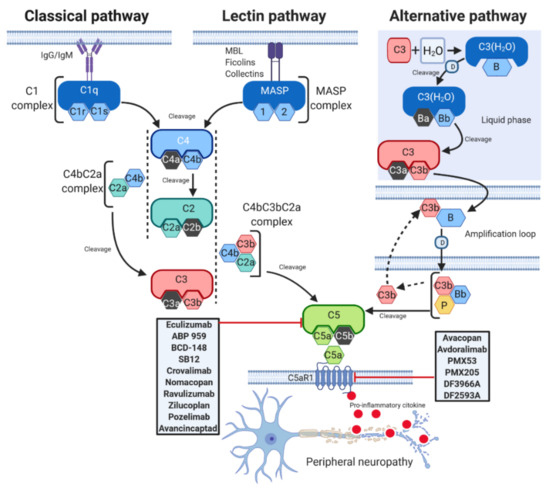
Figure 1.
2
O)) interacts with factor B and factor D to form the fluid phase alternative pathway C3 convertase C3(H2
O)Bb [19]. C3(H2
O)Bb cleaves native C3 to generate C3a and C3b, and the latter binds to its receptor on the lipid membrane. On the membrane, C3b combines with factor B, which is cleaved by factor D to form the alternative pathway C3 convertase C3bBb [14]. Properdin (P), a positive regulator of the complement system, stabilizes C3bBb, and the binding of additional C3b (amplification loop) to the existing alternative pathway C3 convertase generates a C5 convertase, thus leading to the production of C5a and C5b [14].As it is a very complex system, various mechanisms can interfere with the complement cascade leading to over-activation and consequent neuronal damage and disease [20]. Dysregulated complement mechanisms, such as those involving C5 components, contribute to the initiation and progression of several neuropathies [21][22].
As it is a very complex system, various mechanisms can interfere with the complement cascade leading to over-activation and consequent neuronal damage and disease [20]. Dysregulated complement mechanisms, such as those involving C5 components, contribute to the initiation and progression of several neuropathies [21,22].3. Peripheral Neuropathies and C5a/C5aR1 Axis
The term “neuropathy”, or also peripheral neuropathy (PN), refers to a group of conditions characterized by damage and loss of function of nerve cells in the brain or peripheral nervous system (PNS). The population prevalence is about 2400 per 100,000 rising with age to 8000 per 100,000 [23]. Although the damage occurs most frequently in the PNS, also brain injuries, such as stroke, can result in neuropathic symptoms [24]. Moreover, neuropathies can be expressions of neurodegenerative diseases [25], where the degeneration of sensory nerve fibres is due to a wide variety of insults, including diabetes, infectious diseases and nutritional deficiencies, and chemotherapy treatments [26]. Symptoms usually include numbness and paresthesia and are often accompanied by weakness and pain [27][28].
The term “neuropathy”, or also peripheral neuropathy (PN), refers to a group of conditions characterized by damage and loss of function of nerve cells in the brain or peripheral nervous system (PNS). The population prevalence is about 2400 per 100,000 rising with age to 8000 per 100,000 [23]. Although the damage occurs most frequently in the PNS, also brain injuries, such as stroke, can result in neuropathic symptoms [24]. Moreover, neuropathies can be expressions of neurodegenerative diseases [25], where the degeneration of sensory nerve fibres is due to a wide variety of insults, including diabetes, infectious diseases and nutritional deficiencies, and chemotherapy treatments [26]. Symptoms usually include numbness and paresthesia and are often accompanied by weakness and pain [27,28].So far, progress in developing treatments for neuropathies has been frustratingly slow. In fact, despite the availability of therapies that can alleviate symptoms—as, for example, in the case of mild pain, which may be relieved by over the counter analgesics and topical patches—and can address conditions associated with PN [29][30][31][32], no treatments have been approved to date that directly modulate the underlying mechanisms of neuropathies. Current pharmacological therapies are only partially effective, and prolonged exposure to such agents can cause unwanted side effects. Consequently, there is an urgent need to identify and label specific molecular targets and to develop agents to treat pain by exploiting alternative biological pathways.
So far, progress in developing treatments for neuropathies has been frustratingly slow. In fact, despite the availability of therapies that can alleviate symptoms—as, for example, in the case of mild pain, which may be relieved by over the counter analgesics and topical patches—and can address conditions associated with PN [29,30,31,32], no treatments have been approved to date that directly modulate the underlying mechanisms of neuropathies. Current pharmacological therapies are only partially effective, and prolonged exposure to such agents can cause unwanted side effects. Consequently, there is an urgent need to identify and label specific molecular targets and to develop agents to treat pain by exploiting alternative biological pathways.Over the last few years, evidence has indicated that C5a activation triggers a cascade of events that are involved in the pathophysiology of PN and in the genesis of painful states of neuro-inflammation [8]. C5a exerts its biological functions by binding two receptors, C5a receptor-like 1 (C5aR1, also referred to as CD88), a class A seven-transmembrane G-protein-coupled receptor (GPCR), and C5a receptor-like 2 (C5aR2, also known as C5L2 or GPR77) [33], a homolog of C5aR1, but which is not coupled to intracellular heterotrimeric G-proteins due to a mutation in G-protein recognition sequence. C5aR1 is expressed by a broad range of cell types, including all cells of myeloid origin (neutrophils, eosinophils, monocytes, macrophages, dendritic cells, mast cells), lymphocytes, and non-myeloid cells, such as lung, liver, kidney, skin, and central nervous system (CNS) cells [5][34]. C5aR2 is highly expressed in human tissues, such as bone marrow, spleen, and lung, as well as in immune cells, including most myeloid cells and specific T cell subsets [35]. C5aR1 is well-known for its pro-inflammatory effect [36]; conversely, the role of C5aR2 is poorly understood and still controversial [37]. Although C5aR2 can independently induce and modulate C5a biological functions through α-arrestin signalling, further investigations are needed to better understand its actual role [38].
Over the last few years, evidence has indicated that C5a activation triggers a cascade of events that are involved in the pathophysiology of PN and in the genesis of painful states of neuro-inflammation [8]. C5a exerts its biological functions by binding two receptors, C5a receptor-like 1 (C5aR1, also referred to as CD88), a class A seven-transmembrane G-protein-coupled receptor (GPCR), and C5a receptor-like 2 (C5aR2, also known as C5L2 or GPR77) [33], a homolog of C5aR1, but which is not coupled to intracellular heterotrimeric G-proteins due to a mutation in G-protein recognition sequence. C5aR1 is expressed by a broad range of cell types, including all cells of myeloid origin (neutrophils, eosinophils, monocytes, macrophages, dendritic cells, mast cells), lymphocytes, and non-myeloid cells, such as lung, liver, kidney, skin, and central nervous system (CNS) cells [5,34]. C5aR2 is highly expressed in human tissues, such as bone marrow, spleen, and lung, as well as in immune cells, including most myeloid cells and specific T cell subsets [35]. C5aR1 is well-known for its pro-inflammatory effect [36]; conversely, the role of C5aR2 is poorly understood and still controversial [37]. Although C5aR2 can independently induce and modulate C5a biological functions through α-arrestin signalling, further investigations are needed to better understand its actual role [38].By contrast, the C5a/C5aR1 axis triggers leukocyte recruitment and pro-inflammatory cytokines production, which drive inflammatory and neuropathic pain [39][40][41]. Up-regulated levels of C5a and C5aR were found in spinal cord microglia in animals subjected to spared nerve injury (SNI), a model of neuropathic pain [42], while local activation of C5aR1 was found to be implicated in the mechanical nociceptive sensitization in an in vivo model of postoperative pain [43]. In a similar model, PMX-53, a C5aR1 antagonist, decreased mechanical allodynia, oedema, and the levels of several inflammatory mediators present in incised skin [44]. Moreover, local pre-treatment of rats with PMX-53, attenuated mechanical hyperalgesia induced by zymosan, carrageenan, lipopolysaccharide, and ovalbumin, suggesting its role in the control of inflammatory pain [45]. In addition, oral administration of DF2593A, a non-competitive allosteric C5a inhibitor, effectively reduced mechanical hyperalgesia in a carrageenan and complete Freund’s adjuvant-induced inflammatory pain model. Furthermore, DF2593A reduced mechanical hypersensitivity in a model of neuropathic pain induced by SNI [40]. Notably, C5aR1 disruption in knock-out (KO) mice suppressed thermal hyperalgesia compared to wild-type (WT) mice and decreased mechanical sensitization after paw incision [40][41][43], suggesting a major involvement of C5a/C5aR1 axis in pain and inflammation after surgery.
By contrast, the C5a/C5aR1 axis triggers leukocyte recruitment and pro-inflammatory cytokines production, which drive inflammatory and neuropathic pain [39,40,41]. Up-regulated levels of C5a and C5aR were found in spinal cord microglia in animals subjected to spared nerve injury (SNI), a model of neuropathic pain [42], while local activation of C5aR1 was found to be implicated in the mechanical nociceptive sensitization in an in vivo model of postoperative pain [43]. In a similar model, PMX-53, a C5aR1 antagonist, decreased mechanical allodynia, oedema, and the levels of several inflammatory mediators present in incised skin [44]. Moreover, local pre-treatment of rats with PMX-53, attenuated mechanical hyperalgesia induced by zymosan, carrageenan, lipopolysaccharide, and ovalbumin, suggesting its role in the control of inflammatory pain [45]. In addition, oral administration of DF2593A, a non-competitive allosteric C5a inhibitor, effectively reduced mechanical hyperalgesia in a carrageenan and complete Freund’s adjuvant-induced inflammatory pain model. Furthermore, DF2593A reduced mechanical hypersensitivity in a model of neuropathic pain induced by SNI [40]. Notably, C5aR1 disruption in knock-out (KO) mice suppressed thermal hyperalgesia compared to wild-type (WT) mice and decreased mechanical sensitization after paw incision [40,41,43], suggesting a major involvement of C5a/C5aR1 axis in pain and inflammation after surgery. In sum, the administration of C5aR1 antagonists produce analgesic effects in various models of inflammatory and neuropathic pain, highlighting the therapeutic potential of pharmacologically targeting the C5a/C5aR1 axis for chronic pain management.4. Disease of the Peripheral Nervous System (PNS)
The role of C5a/C5aR axis activation in pain generation in neuropathies has been widely investigated in several pharmacological studies. The following paragraphs describe the more recent major findings in the field of PNS and discuss possible implications of the complement system in the pathogenesis of different neuropathic disorders.
4.1. Guillain-Barré Syndrome
GBS is a clinically heterogeneous spectrum of rare post-infectious neuropathies that usually occur in otherwise healthy patients and encompasses acute inflammatory demyelinating polyradiculoneuropathy (AIDP), acute motor axonal neuropathy (AMAN), acute motor-sensory axonal neuropathy (AMSAN), Miller–Fisher syndrome (MFS) and some other regional variants [46][47][48]. GBS is estimated to affect about 1 in 100,000 people each year and it can strike at any age and both sexes [49]. The exact cause of GBS is not known; it is characterized by symptoms that often affect the arms, breathing muscles, and even the face, reflecting widespread nerve damage. Several pathologic and etiologic subtypes of GBS exist, and in many cases it develops subsequently to minor infections but is not associated with other autoimmune or systemic disorders. Usually, GBS occurs after an infectious disease, during which antibodies that cross-react with gangliosides at nerve membranes-with anti-GQ1b ganglioside antibodies being the principal biomarkers of GBS [50]—are aberrantly generated and directed against the PNS, causing nerve damage or impairment of nerve conduction [51]. Anti-GQ1b ganglioside antibodies are principal biomarkers of GBS [50]. The concept of infection-triggered antibody cross-reactivity is well established in axonal GBS and this mechanism is suspected to play a key role in demyelinating GBS. Intravenous administration of immunoglobulins and plasma exchange are effective in treating GBS [52][53]; other therapeutic strategies have been tested in animal models, but their bench-to-bedside transfer is still lacking [54].
Inhibition of C5 complement component activation in experimental ex vivo and in vivo GBS models was extensively used to investigate the pathogenesis of GBS and to evaluate complement deposition in the nerve membrane [55][56][57][58][59]. Specifically, the complement inhibitor APT070 (Mirococept), which regulates C5 and C3 convertases, was shown to be efficacious in an anti-GQ1b-mediated mouse model of the GBS variant MFS, inhibiting the formation of MAC complexes and protecting nerve terminals [57]. Similarly, the anti-C5a monoclonal antibody eculizumab, which inhibits formation of C5a and C5b-9, was reported to prevent complement damage and respiratory paralysis in another severe in vivo mouse model of MFS generated via anti-GQ1b antibody and normal human serum injection as a complement source [58]. Together, these findings have raised the possibility of developing clinical trials using anti-C5a in GBS and in other antibody-mediated terminal motor neuropathies involving complement activation.
4.2. Chronic Inflammatory Demyelinating Polyradiculoneuropathy
Individuals with CIDP lack a detectable antibody titer specific for major compact myelin proteins, thus suggesting that serum constituents, such as cytokines or components of the complement cascade, rather than myelin-directed antibodies might contribute to peripheral nerve injury [60][62][63]. Supporting the hypothesis that complement activation can be a potential pathogenic mechanism for this disease, complement component C3d deposition has been detected on the outer surface of PNS Schwann cells in biopsies from patients with CIDP [60][64][65]. Moreover, clinical studies demonstrated that CIDP patients have increased serum and cerebrospinal fluid levels of C5a [66], which is the result of the proinflammatory function of C3d aimed at recruiting myeloid cells, such as macrophages, to inflammation sites through complement receptors and inducing tissue injury through formation of the MAC. These findings suggest that systemic and local terminal complement activation is a characteristic feature of inflammatory demyelinating polyneuropathies and support a role of complement activation in the pathogenesis of CIDP.
4.3. Familial Amyloid Polyneuropathy
FAP, or transthyretin (TTR) amyloid polyneuropathy, is a progressive sensorimotor and autonomic neuropathy of adult onset, which is characterized by systemic accumulation of amyloid fibrils constituted of aberrant TTR protein [67]. The global prevalence is unknown, but in Japan it has been recently estimated to be around 1 person per million in the general population [68]. FAP is a heterogeneous disorder with a clinical presentation that varies based on the genotype and geographic origin [69][70]. To date, more than 40 TTR mutations have been identified and associated with different patterns of organ involvement, age of onset and disease progression [71][72]. The most common type of mutation is a substitution of valine for methionine at position 30 (ATTRV30M) [73]. The symptoms depend on the site of protein accumulation in the body and, although each TTR variant leads to a different phenotype, PN and cardiomyopathy are predominant hallmarks [74]. The disease usually worsens over 5 to 15 years, and often leads to death caused by heart failure due to TTR protein deposits. Liver transplantation is currently the only treatment for preventing synthesis of the amyloidogenic variants of TTR [75].
Nerve biopsies of individuals with amyloidogenic TTR revealed that in amyloid deposits, transthyretin is aggregated with several other proteins, such as apolipoprotein E, serum amyloid P, and complement C1q [76], suggesting a role for C1q in the pathogenesis of the disease. C1q protein has been shown to be involved also in other amyloidosis, such as Alzheimer’s disease, activating the complement pathway leading to neuronal loss [77]. It is speculated that the complement plays a dual role: although it is known that C1q is able to exert a neuroprotective function against toxic concentrations of soluble pre-amyloid aggregates [78][79], C5a is recognized as having a detrimental neuro-inflammatory effect [80]. However, the impact of C5aR/C5a axis activation in ATTRV30M amyloidosis remains to be clarified.
4.4. Chemotherapy-Induced Peripheral Neuropathy
CIPN is the most common neurologic complication of chemotherapy, often limiting the efficacy of cancer treatments [81]. Between 30% and 40% of patients receiving chemotherapy are reported to experience CIPN, and this number is expected to grow as more aggressive pharmacological agents emerge and survival rates increase [82]. The incidence of CIPN varies from 10% to 100%, depending upon the specific anticancer drug or drug combination administered and upon the dosing regimen [83]. CIPN causes pain, sensory loss and poor dexterity, with a significant impact on patient quality of life. When pain is too severe, a change in chemotherapy regimen may be required, with risk of reducing the therapeutic efficacy, or patients may choose to discontinue the treatment [84]. For example, both oxaliplatin and paclitaxel, two widely used chemotherapeutics, have been shown to cause neurotoxicity and alterations in sensory neurons, triggering CIPN [85][86].
CIPN is the most common neurologic complication of chemotherapy, often limiting the efficacy of cancer treatments [81]. Between 30% and 40% of patients receiving chemotherapy are reported to experience CIPN, and this number is expected to grow as more aggressive pharmacological agents emerge and survival rates increase [82]. The incidence of CIPN varies from 10% to 100%, depending upon the specific anticancer drug or drug combination administered and upon the dosing regimen [83]. CIPN causes pain, sensory loss and poor dexterity, with a significant impact on patient quality of life. When pain is too severe, a change in chemotherapy regimen may be required, with risk of reducing the therapeutic efficacy, or patients may choose to discontinue the treatment [84]. For example, both oxaliplatin and paclitaxel, two widely used chemotherapeutics, have been shown to cause neurotoxicity and alterations in sensory neurons, triggering CIPN [85,86].
Current CIPN management is far from satisfactory, and this is largely due to an inadequate understanding of the complexity of CIPN pathophysiology. The main neurobiological mechanisms involved in CIPN include impaired immune cell signalling and ion channel expression, neurotoxicity, mitochondrial dysfunction, and axon degeneration [87]. Emerging evidence suggests that the immune system and immune-mediated neuro-inflammation are crucial events in the development of CIPN [88]. In particular, a recent study reported that in paclitaxel-induced mechanical allodynia the complement cascade is reduced in C3 KO rats compared to WT animals, and that MAC is tightly involved in the damage of neuronal cells, suggesting that complement may be a novel target for the treatment of CIPN [89]. However, since C3 deficiency almost completely abolishes the release of C5a and MAC, which are crucial for the physiological protection against pathogens, and thus exposes the patient to an increased risk of infections and related side effects, the role of C5a-mediated signalling in CIPN models should be further investigated with the aim to develop more targeted treatment.