Transcytosis of polymeric IgA and IgM from the basolateral surface to the apical side of the epithelium and subsequent secretion into mucosal fluids are mediated by the polymeric immunoglobulin receptor (pIgR).
- polymeric immunoglobulin receptor (pIgR)
- immunoglobulin transcytosis
1. Introduction
The mucosa is an extensive layer of protection for the respiratory, gastrointestinal and urogenital tracts and other secretory glands such as the mammary glands. Separating the internal and external environments, the mucosa is constantly exposed to a wide variety of microorganisms and extrinsic molecules including bacteria, viruses, fungi and toxins. In human beings, the total surface area of the epithelial barrier is about 400 m2 [1]. Protection of the mucosal epithelium is provided by a vast network of proteins, molecules and cells, which are collectively termed as mucosal immunity [2].
Among a myriad of effectors in mucosal immunity, polymeric immunoglobulins IgA and IgM are of particular importance. Immunoglobulin (Ig) is the antigen-recognition molecule derived from B cells, while antibodies are secreted versions of immunoglobulin. An antibody is formed by two identical pairs of heavy and light chains joined together by disulfide bonds. There are five main classes of antibodies: IgA, IgD, IgE, IgG and IgM, which can be distinguished by their heavy chains. Only IgA and IgM can polymerize. The formation of IgA dimers and IgM pentamers is mediated by the joining chain (J chain), while IgM hexamers can be formed in the absence of J chain. Polymerized IgA, and to a lesser extent IgM, protect the mucosal surfaces from infection. In addition, small amounts of IgD are secreted into the mucosal surfaces of oral, nasopharyngeal and lachrymal areas [3]. Daily production of IgA in humans reaches 40 to 60 mg per kg of bodyweight, which is higher than that of all of the other immunoglobulin isotypes combined [4].
Delivery of antibodies to the mucosal surfaces and secretion in milk requires transport across epithelial layers. The polymeric immunoglobulin receptor (pIgR) recognizes the J chain region of polymerized IgA and IgM and transports the antibodies across the epithelial cell. Following proteolytic cleavage of pIgR, polymerized Ig is secreted and released into the luminal space. Since its function was first discovered in the 1980s, J chain, along with the molecular details of Ig polymerization, has largely been overlooked in the field of immunological research. It was only in the last decade when researchers started to take notice of marginal zone B and B1 cell-specific protein (MZB1) [5][6][7][5,6,7], a novel regulator of J chain-mediated Ig polymerization that precedes pIgR-mediated transcytosis.
In this review, we present an overview of how pIgR mediates transcytosis and the consequences of pIgR deficiency. We also expand on the molecular details of J chain binding to Ig polymers and recognition by pIgR based on results from recently published structural studies. We further highlight the proposed roles of MZB1 in the polymerization of IgA and IgM and briefly summarize the latest reports that have implicated MZB1 in human diseases.
2. Polymeric Immunoglobulin Receptor (pIgR)
2.1. Structure and Expression of pIgR
Figure 1). For structures of pIgR in vertebrates other than mammals, there have been several excellent articles covering the expression, structure and functions of pIgR in amphibians [14][15], fish [16][17][18], birds [19] and reptiles [20][21].
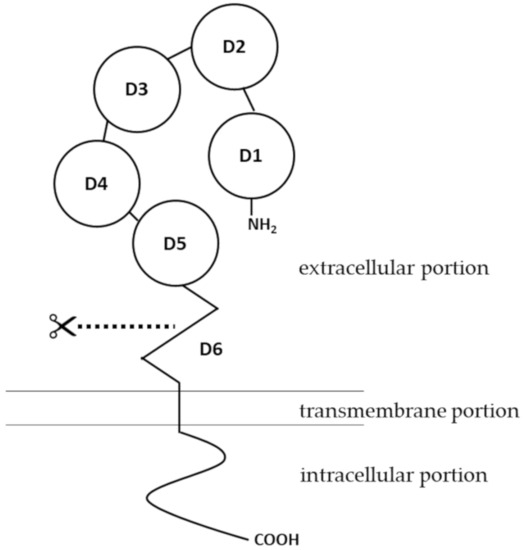
The human
pIgR
pIgR gene spans about 19 kb [22]. PIgR is expressed on epithelial cells of the gastrointestinal tract, respiratory tract and the skin, as well as on the glandular epithelial cells of the breast and liver [1][23][24][25]. A variety of immunological factors have been identified to upregulate expression of pIgR, including interleukin-1 (IL-1), interleukin-17 (IL-17), interferon-γ (IFN-γ), and tumor necrosis factor-α (TNF-α) [26][27][28][29]. Early studies also pointed out that interleukin-4 (IL-4), when acting in synergy with IFN-γ, can upregulate the expression of pIgR [30][31]. The effects of these cytokines on pIgR expression are mediated by transcription factors such as nuclear factor-κ light chain enhancer of activated B cells (NF-κB) and interferon regulatory factor-1 (IRF-1) [32][33], binding sites of which are located in the 5′-flanking region and intron 1 of
pIgR
From a functional perspective, upregulation of pIgR expression levels has been associated with bacterial, viral and chlamydial infections, where the immune system is activated and antibodies are produced and trafficked to fight off the pathogens [35][36][37][38][39]. Some pathogens have evolved strategies to utilize or suppress pIgR expression for the benefit of their infection.
Streptococcus pneumoniae
Candida albicans and Epstein-Barr virus (EBV) can bind to pIgR, which aids their attachment to epithelial cells [40][41][42].
Escherichia coli and simian immunodeficiency virus (SIV) have been reported to downregulate pIgR expression, thereby evading the mucosal immune response [43][44][45]. The commensal microbiome also modulates pIgR expression. It was first reported when colonization of germ-free mice with a commensal bacterial strain
Bacteroides thetaiotaomicron stimulated pIgR expression [46]. Later it was found that pIgR expression could be stimulated in vitro in HT-29 cells, a human intestinal epithelial cell line, when these cells were co-cultured with commensal bacterial strains from the family Enterobacteriaceae [33][47]. It was then proposed that microbial-associated molecular patterns (MAMPs) secreted from the commensal microbiome stimulate epithelial Toll-like receptors (TLRs), which triggers transcription of the
pIgR gene by activating MyD88-dependent signaling pathways [23][25].
Interestingly, studies in mice have linked increased pIgR expression levels in submandibular glands to body exercise and heat acclimatization. Both studies have attributed this phenomenon to mild physiological stress, which might trigger an immune response [48][49]. In the past decade, modulation of
pIgR expression, either elevated or reduced, has been increasingly reported in patients of cancer and metastasis, especially in hepatocellular and pancreatic cases [50][51][52][53][54][55][56]. It is possible that regulation of pIgR expression extends beyond the immune response. Further studies are required to elucidate the underlying mechanisms linking pIgR expression to cancer.
2.2. Functions of pIgR in Transcytosis of IgA and IgM
C
Transcytosis of IgA dimers and IgM pentamers is initiated once they bind to pIgR, and the Ig-pIgR complex is internalized into the cytoplasm of the epithelial cell via clathrin-mediated endocytosis, as was shown in an early in vitro study where pIgR was extrinsically expressed in MDCK cells [61]. Internalization of pIgR can occur even in the absence of its ligand [61]. The internalized Ig-pIgR complex travels along the endosomal pathway. The complex is first trafficked to the basolateral early endosome (EE), followed by transport to the common endosome (CE), before being sorted to the apical recycling endosome (ARE) that is localized beneath the apical epithelial membrane [62]. Sorting and targeting of pIgR throughout the endosomal transcytosis pathway is mediated by a signal of 17 membrane-proximal a.a. residues at the intracellular portion of the pIgR structure [63]. At the apical cell surface, the extracellular portion of pIgR, which binds to polymerized Ig molecules, undergoes endo-proteolytic cleavage at domain 6. The identity of the enzyme responsible for this cleavage remains obscure. The cleaved extracellular portion is referred to as the secretory component (SC). IgA dimers or IgM pentamers, which are originally bound by the SC, are released from the remaining transmembrane and intracellular portions of pIgR. The free, unbound SC-Ig polymer complexes are released as secretory Ig and diffuse into the mucus, where they act as an immunological barrier against infections by denying pathogens access to the epithelium [13][57][64]. This function of secretory Ig has been specifically termed as “immune exclusion” [65].
N-glycosylation, so SC can help localize the secretory Ig complex in the mucus layer [67][68]. Even in the absence of Ig polymers, SC itself may bind and neutralize bacteria and toxins via its glycan moieties [1][69]. Cryo-EM structure of SC complexed with an IgA dimer showed that N65, N72, N168, N403, N451 and N481 are spatially away from any SC-IgA interaction surfaces, so glycosylation at these asparagine residues could be involved in the host and pathogen binding [70]. Functions of SC may be especially important for immunity in breast-fed infants, as the abundance of SC in both its free and Ig-bound forms has long been recorded in maternal milk [71][72][73].
2.3. Consequences of pIgR Deficiency
Studies on genetic knockout mice were pivotal in expanding our understanding of the functions of pIgR. The earliest studies of pIgR−/− mice were conducted in 1999. Epithelial transport of IgA was significantly reduced, although not ablated, in bile, feces and intestinal contents in pIgR−/− mice. Meanwhile, serum IgA levels were markedly increased in pIgR−/− mice [74][75][74,75]. These results demonstrated the essential roles of pIgR in transcytosis of IgA into the intestinal lumen, yet a small amount of IgA may be secreted via other pathways. Since the route for IgA transcytosis into the intestinal lumen is blocked in these genetically-deficient mice, IgA only has access to the blood. The increase in serum IgA levels might be further accounted for by increased numbers of plasma cells that secrete IgA, which was reported in both the lamina propria and the Peyer’s patch of pIgR−/− mice [76][77][76,77]. In addition, a lack of secretory IgA was observed in the pulmonary airways of pIgR−/− mice that developed signs similar to chronic obstructive pulmonary disease (COPD) [78][79][80][78,79,80]. These signs, which were caused by local infection and inflammation, were exacerbated by neutrophils of increased counts and activities [80].
Microbiota in the gut is altered as a result of pIgR deficiency. This was directly confirmed by comparing the intestinal microbiota between pIgR−/− and WT mice using 16s rRNA analysis [81]. Intestinal integrity was mildly compromised in pIgR−/− mice, which might be attributed to a slightly more severe bacterial insult in pIgR−/− mice [82][83][82,83]. This may explain the results from an early study, which showed that pIgR−/− mice were profoundly more sensitive to infection with Salmonella typhimurium via the fecal-oral route, and that bacteria excreted from pIgR−/− mice after S. typhimurium infection were more contagious for other mice [84], as the composition of the excreted bacterial population may differ between pIgR−/− and WT mice.
It is not surprising that pIgR deficiency has been extensively linked to inflammatory diseases in the gut, given the central role of secretory IgA in suppressing inflammation and maintaining homeostasis in the gut [85][86][85,86]. Dextran sulfate sodium (DSS)-induced colitis in mice is the most widely used animal model to study mechanisms of inflammatory bowel diseases (IBD) in humans, which mainly comprise ulcerative colitis and Crohn’s disease [87]. Morbidity and mortality of DSS-induced colitis were significantly enhanced in pIgR−/− mice [81]. Similarly, reduced levels of pIgR and secretory IgA in the gut, as a result of genetic deficiency in IL-17, were correlated with increased weight loss and more severe intestinal inflammation in mice following DSS administration [88]. Lower mRNA levels of pIgR in colonic mucosa have been proposed as a potential biomarker for the clinical diagnosis of IBD [89]. More recently, several cutting-edge studies compared whole-genome sequencing data from the colonic tissues of human IBD patients to those of healthy donors. Among these IBD patients, pIgR has been discovered as one of the most commonly shared sites of somatic mutations that are correlated with impaired protein functions, and these somatic mutations tend to accumulate with age [90][91][92][90,91,92]. Taken together, these data unequivocally illustrate the necessity of pIgR in transcytosis of polymeric immunoglobulins and thus in protection against inflammation.