Porcine reproductive and respiratory syndrome virus (PRRSV) affects the global swine industry and causes disastrous economic losses each year. The genome of PRRSV is an enveloped single-stranded positive-sense RNA of approximately 15 kb. The PRRSV replicates primarily in alveolar macrophages of pig lungs and lymphatic organs and causes reproductive problems in sows and respiratory symptoms in piglets. To date, studies on how PRRSV survives in the host, the host immune response against viral infections, and pathogenesis, have been reported. PRRSV vaccines have been developed, including inactive virus, modified live virus, attenuated live vaccine, DNA vaccine, and immune adjuvant vaccines. However, there are certain problems with the durability and effectiveness of the licensed vaccines. Moreover, the high variability and fast-evolving populations of this RNA virus challenge the design of PRRSV vaccines, and thus effective vaccines against PRRSV have not been developed successfully. As is well known, viruses interact with the host to escape the host’s immune response and then replicate and propagate in the host, which is the key to virus survival.
- PRRSV
- host
- interaction
- immune responses
- vaccines
Note: The following contents are extract from your paper. The entry will be online only after author check and submit it.
1. Introduction
Porcine reproductive and respiratory syndrome virus (PRRSV) has undoubtedly become a substantial financial issue that has affected pig production and caused substantial economic losses for the swine industry worldwide since its outbreak in the 1980s [1][2][1,2]. Annual economic losses in the US due to PRRSV manifested in reproductive problems in sows and respiratory symptoms in piglets were assessed as reaching USD 664 million in 2013 [3][4][3,4]. The latest economic estimate in Germany indicated the losses on farm profits due to the PRRS virus were −19.1% on average and −41% in the worst case [2]. As a global swine pathogen that has caused catastrophic economic losses, PRRSV is the cause for continuing and widespread concern [5].
As a member of the Nidovirales order in the Arteriviridae family, PRRSV is an enveloped virus with an average diameter of 55 nm; it is a positive-stranded RNA virus with an approximately 15 kb genome with a 5′ cap and a 3′ poly A tail [4][6][4,6]. Except for the 5′ and 3′ untranslated regions at both ends, the PRRSV genome contains at least 11 known open reading frames (ORFs) [7]. The first two ORFs occupy about 75% of the viral genomes coding for polyproteins, including pp1a and pp1ab, by ribosome shifting, and then the PRRSV proteases hydrolyze and cleave polyproteins into at least 16 distinct nonstructural proteins (nsps) [4][8][4,8]. The nsps that participate in viral genome replication and transcription are essential for the survival of the PRRSV [9]. PRRSV virions are composed of an N protein (nucleocapsid protein) and a lipid envelop (GP2, E, GP3, GP4, M, GP5, ORF5a) that envelops the membrane [10][11][10,11]. M and GP5 are the major components of the virus coat [12].
The PRRSV is mainly divided into two genotypes: type 1 (represented by the European strain Lelystad Virus) and type 2 (represented by the North American strain VR-2332), and both PRRSV genotypes have only 50–60% nucleotide identity [6][13][6,13]. In addition to the genotype differences between PRRSV-1 and PRRSV-2, the host immune responses have been shown to differ, sometimes considerably due to their biological differences including pathogenicity (Table 1). A large number of studies have found a general consensus that PRRSV-2 causes more severe respiratory disease than PRRSV-1 [14]. Therefore, it is necessary to emphasize the host immune response induced by different genotypes. Due to high inter-strain genetic exchange and rapid mutations of PRRSV, it has always been a substantial challenge to design effective vaccines and drugs [4].
Table 1.
| Host | PRRSV | Function | Virus Genotypes | References | |||||
|---|---|---|---|---|---|---|---|---|---|
| Heparan | M/GP5 | Concentrate virions on the cell surface | PRRSV-1/PRRSV-2 | [15] | |||||
| pSn | M/GP5 | PRRSV attachment and internalization receptor via clathrin-mediated endocytosis | PRRSV-1 | [16] | |||||
| CD163 (5JFB) | GP2/GP3/ GP4/GP5 |
Uncoating and genome release | PRRSV-1/PRRSV-2 | [13][17] | [13,17] | ||||
| CD151 | 3′ UTR RNA | Cooperate in infection | PRRSV-2 | [18] | |||||
| Simian vimentin | N (1P65) | Mediate transportation of the virus in the cytosol | PRRSV-2 | [19] | |||||
| CD209 | GP5 | Essential in PRRSV entry and infection | PRRSV-1/PRRSV-2 | [20] | |||||
| MYH9 | GP5 | Essential in PRRSV entry and infection | PRRSV-1/PRRSV-2 | [21][22] | [21,22] | ||||
| IFN-β | nsp1α (3IFU) | Suppress IFN by degrading CBP | PRRSV-2 | [23][24] | [23,24] | ||||
| nsp1β (3MTV) | Suppress IFN by inhibiting both IRF-3 and NF-κB-dependent gene induction | PRRSV-2 | [25] | ||||||
| N | Suppress IFN by inhibiting the phosphorylation and nuclear translocation of IRF3 | PRRSV-2 | [26] | ||||||
| nsp2 | Suppress IFN by inhibiting the activation of the IRF-3 and NF-κB signaling | PRRSV-2 | [27] | ||||||
| nsp4 (5Y4L) | Suppress IFN by blocking NF-κB activation | PRRSV-2 | [28] | ||||||
| nsp11 (5EYI) | Suppress IFN by inhibiting the activation of the IRF-3 and NF-κB signaling, and inhibiting the formation and nuclear translocation of ISGF3 targeting IRF9 |
PRRSV-2 | [29][30][31] | [29,30,31] | |||||
| IL-1β | E | Increase the release of IL-1β | PRRSV-2 | [32] | |||||
| nsp11 | Inhibit the secretion of IL-1β | PRRSV-2 | [33] | ||||||
| IL-10 | N | Up-regulate IL-10 via NF-κB and p38 MAPK pathways in PAMs | PRRSV-2 | [34] | |||||
| nsp1 | Up-regulate IL-10 | PRRSV-2 | [35] | ||||||
| Gp5 | Up-regulate IL-10 through p38 MAPK and signal transducer and activator of transcription-3 (STAT3) activation | PRRSV-2 | [36] | ||||||
| IL-17 | nsp11 | Induced IL-17 production depending on the PI3K-p38MAPK-C/EBPβ/CREB pathways | PRRSV-2 | [37] | |||||
| TRIM25 | N | Competitively interact with TRIM25, thereby interfering with TRIM25-mediated RIG-I ubiquitination | PRRSV-2 | [38] | |||||
| TRIM22 | N | Interact with TRIM22 thereby reducing virus replication | PRRSV-2 | [39] | |||||
| TRIM59 | nsp11 | Interact with TRIM59 thereby reducing virus replication | PRRSV-2 | [40] | |||||
| MiR-181 | ORF4 | Inhibit PRRSV replication | PRRSV-2 | [41][42] | [41,42] | ||||
| MiR-130 | 5′ UTR | PRRSV-2 | [43] | ||||||
| MiR-23 | ORF3 | PRRSV-2 | [44] | ||||||
| MiR-378 | ORF7 | PRRSV-2 | [44] | ||||||
| MiR-505 | ORF3/ORF5 | PRRSV-2 | [44] | ||||||
| PIAS1 SLA-I(5YLX) |
nsp1α | Modulate degradation via SUMO E3 ligase activity | PRRSV-2 PRRSV-2 |
[45] [45] | [45 | [46 | ] [45 | ] | ,46] |
| Nup62 | nsp1β | Inhibit host antiviral protein expression | PRRSV-1/PRRSV-2 | [47] | |||||
| STAT3 | nsp5 | Promote the degradation of STAT3 and interference with the JAK/STAT3 signaling | PRRSV-1/PRRSV-2 | [48] | |||||
| pRb | nsp9 | Benefit the replication of PRRSV | PRRSV-2 | [49] | |||||
| NLRX1 | nsp9 | Restrict PRRSV replication | PRRSV-2 | [50] | |||||
| ZAP | nsp9 | Repress PRRSV replication. | PRRSV-2 | [51] | |||||
| Fibrillarin Nucleolin Poly(A)-binding |
N | Function remains to be further clarified | PRRSV-2 | [52] |
2. The Process of the PRRSV Entry and Infection
Numerous studies have been performed to learn more about the biochemistry of the PRRSV. The process of the virus entry into its host cell is the first crucial step in the infection and the focus of basic research [53]. PRRSV has a very narrow cell tropism, and the primary target cells are porcine alveolar macrophages (PAMs) [12][54]. Numerous studies have found that the existence of the specific entry mediators in the target cell leads to its restricted cell tropism [53][55]. These cellular factors, including heparan sulphate, CD163, porcine sialoadhesin (pSn), vimentin, CD151, CD209 (DC-SIGN), non-muscle myosin heavy chain 9 (MYH9), and others, are involved in virus binding, internalization, and genome release, as shown in
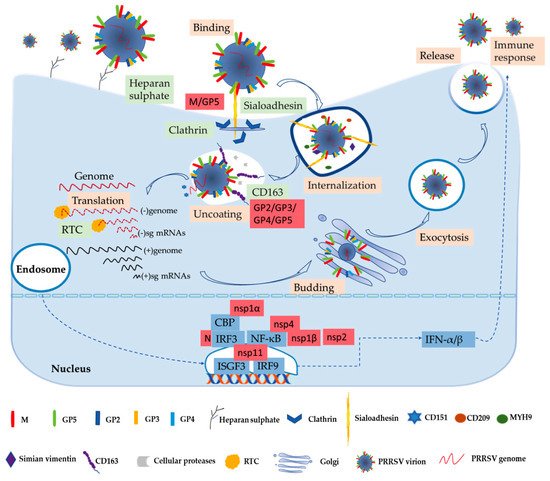
Figure 1.
In the early stages, PRRSV enters the macrophage via a receptor-mediated method [53][57]. PRRSV concentrates virions on the cell’s surface by interacting with heparan sulphate glycosaminoglycans that are present on the surface of mammalian cells, hence promoting a more efficient attachment for subsequent binding to one or more receptors involved in virus internalization; studies have found that PRRSV-1 and PRRSV-2 have different sensitivities to heparin [15][53][58]. Next, the integration with pSn gradually increases. The pSn, a PAMs-restricted type 1 transmembrane glycoprotein, is identified as a PRRSV attachment and internalization receptor via clathrin-mediated endocytosis [53][58]. The heparan sulphate enhances the interaction between the virus and sialoadhesin, but is not necessary, and the M/GP5 complex is involved in interacting with heparan and the N-terminal domain of pSn [15][16]. Upon internalization with the participation of pSn, the genome of the virus that is present in the early endosome is released into the cytoplasm [53]. The last stage is critically dependent on CD163, a scavenger receptor cysteine-rich (SRCR) family for hemoglobin clearance, which is the most specific and indispensable receptor for PRRSV entry and infection both in vitro and in vivo [8][12][59]. Additionally, the viral GP2 and GP4 glycoproteins bind to CD163 [13]. Moreover, a study has shown that CD163 interacts with GP3 and GP5 in addition to the known interactions with GP4 and GP2, and co-immunoprecipitation (co-IP) analysis indicated that the SRCR5-domain deletion of CD163 loses its interaction with viral GP2, GP3, and GP5, and thus blocks virus uncoating in the early endosomes [17]. A growing body of research has also indicated that CD163 plays an essential role in viral uncoating and genome release, and CD163 knockout pigs are resistant to PRRSV infection [13][59][60][61]. In addition to CD163, cellular proteases, such as the aspartic protease cathepsin E and trypsin-like serine proteases, have been implicated in the uncoating process and subsequent infection [62]. Moreover, there are some mediators involved in the entry process of PRRSV into the macrophage. CD151 interacting with the PRRSV-2 3′ untranslated region (UTR) RNA should cooperate in infection in vitro [18]. Simian vimentin bound to PRRSV-2 nucleocapsid protein is involved in mediating transportation of the virus in the cytosol [19]. CD209 (DC-SIGN) and MYH9 interacting with GP5 are essential factors in both PRRSV-1 and PRRSV-2 entry and infection [20][21][22].
The genome released by the virus was identified as the template used to translate into pp1a and pp1ab, and after that the proteases hydrolyze and cleave polyproteins into mature nonstructural proteins as described above [63]. The viral replication and transcription complex (RTC) is assembled by nsps, whose key components are nsp9 and nsp10 [64]. The RTC first engages in producing both full-length and subgenome (sg)-length minus strands using a mechanism of discontinuous transcription, the latter serving as templates for the synthesis of plus-strand sg mRNAs required to express the structural protein genes, which reside in the 3′-proximal quarter of the genome [8][65][66]. The last stage is assembling and releasing the virion. Novel generated RNA genomes are packaged into nucleocapsids that become enveloped virions by structural proteins through budding from smooth intracellular membranes, and the new virions are released from the cell in the form of exocytosis [8][65]. The entire process is illustrated in
3. The PRRSV–Host Interactions
The complex networks of virus–host interactions are essential in the overall process of PRRSV entry and infection. The viral structural proteins interact with host receptors to mediate viral entry as described above. The interactions of host and viral nonstructural proteins can exert an influence on the replication and transcription of viral genomes. The virus’s invasion causes a series of immune responses, and then virions escape from the host immune system to favor their own replication by interacting with the host. Only by better understanding the molecular mechanism of PRRSV immune evasion and modulation can we design more effective vaccines. Further studies of these host–virus interactions are critical to the development of novel antiviral strategies against PRRSV.
3.1. Interferons (IFNs)
Figure 1 [13][67]. PRRSV-2 is sensitive to type I interferons (IFN-α/β), and has some immunosuppressive mechanisms of suppressing IFNs [13][68]. Activation of IFN regulatory factor 3 (IRF3) and nuclear factor-κB (NF-κB) plays an important role in activating the IFN-β promoter, and these factors bind to the IFN-β promoter to form an enhanceosome via the cAMP response element-binding (CREB)-binding protein (CBP) transcriptional co-activator [23]. This also suggests that the virus has evolved to assuage the host’s innate immunity [13]. The mechanism of suppressing IFNs by blocking the activation of the IRF3 or NF-κB is an important strategy to respond to the innate immunity of the host to the virus’s proteins, such as nsp1, nsp2, and nsp4 [27][28]. Some studies have revealed that IFNs are suppressed by the PRRSV-2′s nsp1, which is a potent IFN antagonist [24]. The zinc finger 1 motif of nsp1α is essential in suppressing IFNs by inducing CBP degradation to inhibit the recruitment of CBP for enhanceosome assembly, which is likely the key mechanism in IFN suppression [23][24]. Moreover, nsp1β inhibits both IRF3- and NF-κB-dependent gene induction via dsRNA and the Sendai virus, resulting in IFN suppression [25]. Similarly, the PRRSV-2 nucleocapsid (N) protein inhibits the phosphorylation and nuclear translocation of IRF3 to suppress IFNβ induction [26]. Although nsp11 endoribonuclease (NendoU) activity inhibits IFNβ by suppressing IRF3 and NF-κB activation, binding to IRF9 controls the formation and nuclear translocation of the IFN-stimulated gene factor 3 (ISGF3) and antagonizes type I IFN signaling in a NendoU activity-independent manner [29][30][31].
3.2. Interleukin (IL)
In highly pathogenic (HP) PRRSV-infected swine, the cytokines including interleukin (IL)-1, IL-6, IL-8, IL-10, and tumor necrosis factor (TNF)-α are significantly increased [69][70]. The PRRSV-2 envelope protein E remarkably increases the release of IL-1β from lipopolysaccharide (LPS)-primed PAMs [32]. Studies showed that nsp11 NendoU activity plays a key role in inhibiting the secretion of IL-1β [33]. IL-10 is a significant immunosuppression cytokine with anti-inflammatory properties that can counteract adaptive immunity, thereby preventing impairment to the host [71][72]. A previous study has demonstrated that the PRRSV-2 N protein can up-regulate IL-10 via NF-κB and p38 mitogen-activated protein kinase (MAPK) pathways in PAMs [73]. Additionally, in porcine monocyte-derived dendritic cells (MoDCs) and PAMs, the N protein can promote the expression of IL-10 [34]. The GP5 could significantly increase IL-10 production through p38 MAPK and signal transducer and activator of transcription-3 (STAT3) activation [36]. Similarly, nsp1 can increase the level of IL-10 as an inducer in vivo [35]. IL-17 is a proinflammatory cytokine associated with intense inflammation and is upregulated by HP-PRRSV of genotype 2 infection [13][37]. A study found that HP-PRRSV nsp11 could induce IL-17 production depending on the phosphatidylinositol 3-kinase (PI3K)-p38MAPK-C/EBPβ/CREB pathways [37].
3.3. Tripartite Motif (TRIM) Proteins
Tripartite motif (TRIM) proteins, as critical components of the innate immune system, play significant roles in fighting virus invasion for mammalian cells, and can regulate multiple cellular processes including transcription-dependent antiviral responses such as cytokine-mediated or autophagy-mediated antiviral modulation [40][74][75]. The N-terminal RING-type zinc finger domain of TRIM proteins confers E3 ubiquitin ligase activity [74]. Study has indicated that the PRRSV-2 N protein can antagonize the antiviral activity of TRIM25 and suppress innate immune responses of the host by competitively interacting with TRIM25, thereby interfering with TRIM25-mediated retinoic acid-inducible gene I (RIG-I) ubiquitination and inhibiting IFN-β production [38]. TRIM22 can interact with the PRRSV-2 N protein and reduce virus replication, and the SPla and the RYanodine Receptor (SPRY) domain and nuclear localization signal of TRIM22 are indispensable for this interaction [39]. Moreover, the N-terminal RING domain of TRIM59, which is an important antiviral component, can interact with the C-terminal NendoU domain of nsp11, thereby inhibiting PRRSV-2 infection [40].
3.4. MicroRNAs (miRNAs)
MicroRNAs (miRNAs) are small non-coding RNA molecules containing about 21 nucleotides in length, and they play key roles in the complex networks of PRRSV–host interactions [76][77]. More recently, miRNAs have been considered as crucial post-transcriptional gene regulators for viral replication and host immune responses in the process of PRRSV invasion [13][77][78]. This research had indicated that MicroRNA can target signaling pathways or host factors both related to PRRSV replication [5]. MiR-30c has a negative effect on the IFN-I response by targeting Janus kinase 1 (JAK1) to facilitate HP-PRRSV of genotype 2 infection [79]. On the contrary, miR-181 can suppress PRRSV-2 infection by targeting and down-regulating the PRRSV-2 receptor CD163 [41]. MiR-10a-5p can interact with 3′ UTR of pig SRP14 mRNA and reduce SRP14 expression through translational repression, thereby inhibiting PRRSV-1 replication [80]. Additionally, miRNAs also can target the PRRSV genome directly, and this is a new perspective on controlling PRRSV infection [81]. In addition to targeting the 3′ UTR of CD163 mRNA, MiR-181 can combine with ORF4, which is a highly conserved region of PRRSV-2 genomic RNA, and thus inhibits PRRSV-2 replication [41][42]. MiR-130 can directly target the HP-PRRSV 5′ UTR and exert antiviral activity. Further study has reported that miR-130 has an effect on inhibiting the replication of PRRSV-2 strains, but not the replication of classical PRRSV-1 strains [13][43]. MiR-23 and miR-378, which were identified as critical inhibitors of PRRSV-2 replication, can directly target PRRSV genomic ORF3 and ORF7, respectively [44]. Interestingly, miR-505, also as a critical inhibitor of PRRSV-2 replication, can directly target both ORF3 and ORF5.
3.5. Other Host Factors’ Interactions with PRRSV
PRRSV depends on host factors to complete genome replication and interacts with host molecules for its survival and reproduction. The host immune system will take some measures to suppress the virus replication during virus invasion. Besides the abovementioned host factors, other cellular components are also involved in interacting with PRRSV. The cellular protein nucleoporin 62 (Nup62) interacts with nsp1β, leading to inhibition of host antiviral protein expression, revealing a new strategy of immune escape [47]. In addition, research found that cellular retinoblastoma protein (pRb) interacts with the nsp9 of genotype 2 PRRSV, which will benefit the replication of PRRSV-2 [49]. Inversely, the leucine-rich repeats (LRR) domain of nucleotide-binding oligomerization domain-like receptor (NLR) X1 as a new host restriction factor interacts with the RdRp domain of PRRSV-2 nsp9, which restricts PRRSV-2 replication [50]. Similarly, the interaction of the zinc finger domain of ZAP, a zinc finger antiviral protein, and nsp9 mapped to amino acids 150 to 160, can repress PRRSV-2 replication [51]. Nsp1α can combine with the protein inhibitor of activated STAT1 (PIAS1) as the small ubiquitin-related modifier (SUMO) E3 ligase leading to the nsp1α sumoylation [45]. In virtue of the SUMO E3 ligase activity, nsp1α interacts with swine leukocyte antigen class I (SLA-I) to modulate degradation, in the same way facilitating the ubiquitinylation of CBP for degradation [45][46][45,46]. The nsp5 of PRRSV-1 and PRRSV-2 was shown to promote the degradation of signal transducer and activator of transcription 3 (STAT3), a pleiotropic signaling mediator of numerous cytokines, leading to interference with the JAK/STAT3 signaling and the host immune responses [48]. The PRRSV-2 N protein interacts with RNA-associated nuclear host proteins such as fibrillarin, nucleolin, and poly(A)-binding protein, but the specific function remains to be further clarified [52]. Furthermore, the PRRSV N protein appears to up-regulate NF-κB activation that is attributed to the N protein nuclear localization signal [45].
In conclusion, a complex network of PRRSV–host interactions exists throughout the virus cycle, including virus entry, replication, and infection, as shown in Table 1. The immune response caused by different virus genotypes is emphasized in Table 1. Although significant advancements have been made, the understanding of direct or indirect virus–host interaction networks remain limited. Moreover, some seemingly contradictory results are hard to explain. Study indicated that the NF-κB is stimulated by the N protein, which may up-regulate IFN [45]. However, the N protein inhibits the phosphorylation and nuclear translocation of IRF3 to suppress IFNβ induction [26]. The stimulation of NF-κB by the N protein may involve other cellular pathways, and the mechanism of cytokines’ regulation by the N protein needs to be further explored. The interaction of PRRSV nsp9, a critical component of the viral RTC, and some host proteins can inhibit virus replications such as NLRX1 and ZAP, and yet they can promote virus replications such as pRb, which has aroused much concern.