Systemic lupus erythematosus (SLE) is a chronic, systemic autoimmune disease with a relapsing–remitting course and characterized by the production of a wide range of autoantibodies.
- systemic lupus erythematosus
- airway disease
- interstitial lung disease
- shrinking lung syndrome
- diffuse alveolar hemorrhage
- pleurisy
- infection
1. Introduction
Although people of any age and gender can be involved, females of childbearing age are the most affected, with a female-to-male ratio of about 9:1 [1].
SLE can have a wide range of manifestations, involving virtually every organ or apparatus, and its severity can vary from very mild disease without major organ involvement, to severe life-threatening conditions. Clinical manifestations may include cytopenia, fever, malar and other skin rashes, oral ulcers, polyarthralgia/non erosive arthritis, vasculitis, renal, neurological, cardiac and pleuro-pulmonary involvement [2,3,4][2][3][4]. Recently, a new set of classification criteria was proposed by American College of Rheumatology/European League Against Rheumatism (ACR/EULAR), designed to increase classification sensitivity and specificity for inclusion in SLE research studies and trials [5]. Furthermore, recommendations on disease management from EULAR were recently updated [6,7][6][7].
SLE pathogenesis is multifactorial and not completely understood, and includes an interaction between non-Mendelian genetic predisposition, hormonal and environmental factors, ultimately leading to an alteration in both innate and adaptive immunity. In particular, SLE pathogenesis is characterized by an impaired apoptotic cell clearance by phagocytes, B-cell and T-cell autoreactivity leading to an abnormal production of autoantibodies, and immune complexes (ICs) formation with nuclear and cytosolic antigens. ICs can, in turn, activate the classical pathway of the complement system contributing to inflammation and damage in target organs [4,8][4][8].
Although the exact prevalence is unknown, respiratory tract involvement can be present in 50–70% of SLE patients, being the presenting symptom of the disease in 4–5% of cases and more frequent in men [8,9,10][8][9][10]. Every part of the respiratory tract can be involved: upper and lower airways, vessels, pleura, lung parenchyma and respiratory muscles (Figure 1). Respiratory manifestations can be acute or chronic, primary (directly caused by the disease) or secondary (due to concomitant complications such as infections). Interestingly, acute manifestations may be associated with generalized lupus disease activity, while chronic complications may progress independently to general disease activity [10].
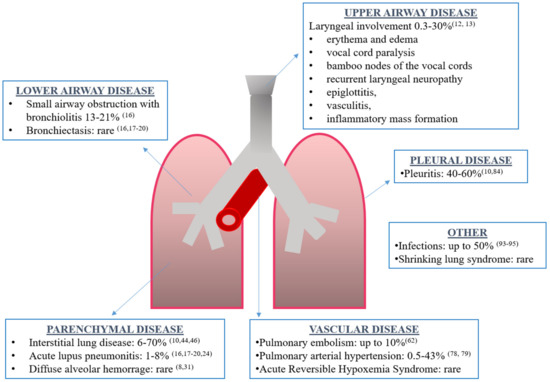
Figure 1. Overview of respiratory manifestations in systemic lupus erythematosus along with the prevalence and corresponding references.
Respiratory manifestations of SLE are associated with a variable mortality rate, depending to the type of involvement, its extension, and the presence of comorbidities. In particular, pulmonary involvement is associated with higher mortality and with negative effect on patient-reported outcomes, patient-performed outcome and quality of life [11]. Unfortunately, clinical and therapeutic trial data specifically focused on respiratory manifestations of SLE are scarce, so treatment options are based on evidence from other organ involvement in SLE, or from respiratory manifestations in other autoimmune diseases, or based on case reports or small cases series.
2. Airway Disease
Laryngeal involvement can occur in 0.3–30% of SLE patients and range from asymptomatic to severe life-threatening upper airway obstruction [13][12]. Clinical manifestations are non-specific and include hoarseness, cough, dyspnea, and stridor. Mucosal inflammation with erythema and edema is the major manifestation; other findings include vocal cord paralysis, bamboo nodes of the vocal cords, recurrent laryngeal neuropathy, epiglottitis, rheumatoid nodules [14][13], vasculitis, inflammatory mass formation and late subglottic stenosis. It usually responds well to corticosteroids (CS) therapy. However, in severe cases of respiratory failure, advanced airway management may be necessary [13,15,16][12][14][15].
Other airway involvement includes upper airway angioedema, necrotic tracheitis and early post-intubation stenosis, bronchial stenosis; small airway obstruction with bronchiolitis is found in the 13% to 21% of patients with the use of high-resolution computed tomography (HRCT) [17][16] and bronchiectasis as a consequence of direct SLE involvement or as sequelae of bronchopulmonary infections [17,18,19,20,21][16][17][18][19][20].
3. Parenchymal Lung Disease
3.1. Acute Diseases
Acute lupus pneumonitis (ALP) and diffuse alveolar hemorrhage (DAH) are acute and uncommon manifestations of SLE [10].
3.1.1. Acute Lupus Pneumonitis
ALP is a rare, probably under-recognized, manifestation of SLE that occurs in 1–8% of SLE patients, in particular younger patients and patients with a recent diagnosis. Moreover, it can be the first manifestation of a previously unrecognized SLE in 50% of cases [10,17,23,24,25][10][16][21][22][23]. Clinical presentation is non-specific and can simulate infectious pneumonia with sudden onset of fever, cough, dyspnea, pleuritic chest pain and occasionally hemoptysis. Physical examination can reveal tachycardia, tachypnoea hypoxemia, hypocapnia and lung crackles. Occasionally, it can present with acute respiratory failure requiring mechanical ventilation. ALP has been described complicating SLE during pregnancy [10,17,23,24,25,26][10][16][21][22][23][24]. Chest X-ray can show multiple, bilateral patchy infiltrations, predominantly in the lower lobes, with or without pleural effusion. However, chest X-ray can be normal, especially in the initial phases or shows only lung nodules. Although these findings are non-specific, CT scan can show ground glass opacities and areas of consolidation, predominantly in the lower lobes [10,23][10][21]. Histologically, ALP presents diffuse alveolar damage (DAD) with inflammatory cell infiltration, damage and necrosis of alveolar-capillary unit, edema, hyaline membrane formation and alveolar hemorrhage. Capillaritis and thrombosis have also been described. Alveolar damage may be mediated by the deposition of ICs and complement fractions. However, there are not diagnostic and/or pathognomonic findings specific for ALP.
3.1.2. Diffuse Alveolar Hemorrhage
DAH, first described by Dr. William Osler in 1904, is a rare, but very severe and potentially fatal complication of SLE [8,32][8][25]. It is not exclusive to SLE, occurring in several other conditions such as anti-neutrophil cytoplasmic autoantibody (ANCA)-associated vasculitis, antiphospholipid syndrome (APS), other connective tissue diseases, infections, bone marrow transplantation, and drug toxicity [33,34][26][27].
DAH prevalence among SLE patients ranges from 0.5–0.6% to 5.4–5.7% with a femal-to-male ratio of approximately 6:1. DAH was described as initial manifestation of SLE in 11–20% of cases; some autoptic studies in SLE patients have found the presence of red blood cells in the lungs of 30–66% of cases maybe due to the presence of either unidentified or subclinical, paucisymptomatic forms of DAH [10,33][10][26]. Mean age of presentation is 27 years, but it can occur at an early stage of the disease [17][16]. Some patients may have recurrent episodes [33,35][26][28].
3.2. Chronic Diseases
Chronic interstitial lung disease (ILD) in SLE seems to be less frequent in comparison to other connective tissue diseases (CTDs), and it is rarely severe [10][29][30][31]. The exact prevalence is probably underestimated, because older studies performing chest X-ray have shown the presence of ILD in 6–24% of SLE patients, while in those using a more sensitive method such as HRCT, ILD was found in up to 70% of cases, suggesting that the condition is frequently subclinical [10][30][32]. Risk factors for ILD include older age, late-onset SLE, illness duration (≥1 year), tachypnea, low levels of anti-dsDNA, high level of C3 and male gender [29][30][31][32][33]. The presence of Raynaud’s phenomenon, swollen fingers, sclerodactyly, telangiectasia, nailfold capillary abnormalities among SLE patients was associated with a higher prevalence of restrictive deficit and reduced DLCO, probably in the context of overlap syndromes that seem to carry a worse lung prognosis. Some associations were found with anti-U1 RNP, anti-SSB, anti-Scl70 and anti-SSA antibodies and sicca syndrome [10][30][31][32][33][34].
The most common pattern, histologically and radiologically, is non-specific interstitial pneumonia (NSIP); however, usual interstitial pneumonia (UIP) is not uncommon [33]. Lian et al. reported that the most frequent findings were ground glass opacities (84.4%), followed by consolidation (21.1%), honeycombing (15.6%), and traction bronchiectasis (12.8%) [34].
Clinically, ILD can evolve as a consequence a disease with acute onset (ALP or DAH) or follow a more insidious onset with chronic non-productive cough, exertional dyspnea and non-pleuritic chest pain. The mean age of onset is earlier when following an acute condition (mean 38 years) compared to the chronic form (46 years). Patients with a radiologically documented ILD can also be asymptomatic [10][32]. Inspiratory fine crackles may be heard upon physical examination, while the presence of digital clubbing is rare. Pulmonary function tests can show a restrictive pattern with reduced DLCO [10]. The severity of ILD does not correlate with SLE serologic markers [30].
Prognosis for SLE-associated ILD seems more favorable when compared to idiophatic pulmonary fibrosis or RA-associated ILD [31][33][35][36]. Toyoda et al. found a five-year survival rates of 92.9% calculated from the time ILD was diagnosed and the survival rate did not significantly differ between the patients with and without ILD [33].
Lymphocytic interstitial pneumonia (LIP) can complicate many autoimmune conditions and has been described in SLE patients in particular when associated with Sjögren’s Syndrome. LIP is characterized by the formation of lung cysts, an infiltration of the interstitium with polyclonal lymphocytes and lymphocytic alveolitis [10][30][37][38]. Prognosis is variable. Approximately 50–60% of patients respond to corticosteroids with stabilization or improvement of the disease, but in others there is progressive decline in pulmonary function and development of honeycomb lung. In general, death occurs in approximately 33 to 50% of patients within 5 years of diagnosis [37][38].
Organizing pneumonia (OP) has also been described as initial manifestation of SLE and regardless of SLE activity [10][30][39][40][41]. On HRCT, OP shows ground glass opacities, consolidations and peribronchovascular opacities. OP has also been described in rhupus syndrome [42]. CS are the treatment of choice. In the majority of cases patients recover within days of weeks after treatment introduction and radiographic findings show improvement in 50–86% of patients. Spontaneous resolution may occur. However, in a minority of cases, the disease may persist, and up to 30% may have a relapse after treatment withdrawal [43]. Several immunosuppressant agents, such as azathioprine, MMF, cyclosporin, CYC and plasmapheresis, have been used in various case reports. [39][40][41][42][43]. Finally, an association between SLE and pulmonary sarcoidosis has been described [10][44][45][46][47]. According to Rajoriya N et al., patients with sarcoidosis have an OR of 8.33 (2.71 to 19.4) for the development of SLE [45].
Placebo-controlled trials to guide the treatment of SLE-associated ILD are lacking. CS are, generally, the mainstay of treatment and patients usually show a good response. Immunosuppressants such as CYC, azathioprine, or MMF can be added in refractory more severe cases [10][21]. Among biologics, RTX can be used in some cases [48].
Treatments are generally well tolerated; with CYC, immuno- and myelosuppression, as well as IgG levels decreased can occur with subsequent infections that are generally non-life-threatening and do not necessitate stopping treatment [49][50]. In particular, in the study of Okada et al., only two sessions of CYC infusions among a total of 141 were postponed because of upper respiratory infections [50]. Interestingly, cumulative data show a higher frequency of adverse events, including hemorrhagic cystitis, premature ovarian failure, herpes zoster and cancer, with the oral administration, in comparison with pulse intravenous infusion of CYC, as found in the lupus nephritis [49][50][51]. Concerning the use of MMF in SLE-ILD, only one of ten patients with CTD-ILD had a diagnosis of SLE in the case series by Saketkoo et al. [52], while Fisher et al. included four patients with SLE-ILD in their retrospective study [53]. The most common side effects reported in these studies were diarrhea and leucopenia.
4. Vascular Diseases
4.1. Acute Reversible Hypoxemia Syndrome
First described in 1991 by Abramson [73][54], acute reversible hypoxemia syndrome is characterized by the acute onset of dyspnea, chest pain and hypoxemia. Pleural involvement may be present. It is frequently associated with a flare of SLE. Pulmonary imaging is generally normal, while PFTs may show reduction in vital capacity and DLCO [17,51][16][32]. Pathophysiology is not completely understood. An association between endothelium activation, with a high expression of vascular adhesion cell molecule-1 (VACM-1) and intercellular adhesion molecule-1(ICAM-1), and activated neutrophil and platelet sludging mediated by complement activation has been postulated as a pathogenic mechanism. These alterations can ultimately lead to endothelial dysfunction, vascular lumen occlusion by leukocyte aggregates and subsequent hypoxemia [17,51,73,74][16][32][54][55].
This condition rapidly responds to low doses of CS, usually insufficient to control SLE flares, when present together, so higher doses may be needed. Combination of high doses of aspirin can be useful [17[16][32],51], and most cases respond to therapy with rapid improvement of gas exchanges [9].
4.2. Pulmonary Embolism
SLE patients are at increased risk of developing deep vein thrombosis (DVT), occurring in up to 10% of patients [75][56], and pulmonary embolism (PE) with a 3-fold increased risk in comparison to general population [76][57]. Vein thromboembolism (VTE) represents the third most common cardiovascular (CV) event after myocardial infarction and stroke [77,78][58][59]. PE has a high mortality rate of up to 15%. Many risk factors have been investigated besides “classical” risk factors such as obesity, hyperglycemia and hyperlipidemia [77][58]. Moreover, You et al. found the following risk factors associated with PE: high body max index, hypoalbuminemia, positivity for anti-phospolipid antibodies (aPL), high levels of high sensitivity CRP and high doses of CS (>0.5 mg/kg/day) [78][59]. Finally, SLE patients with APS are at increased risk of DVT and PE. The prevalence of APS among SLE patients is about 30% [79][60].
APS can cause a hypercoagulable state by interacting and activating platelets, neutrophils and endothelial cells [78][59]. In particular, a metanalysis found that SLE patients with APS have a six times greater risk of developing PE than SLE patients without APS [79][60]. Moreover, patients with the positivity for lupus anticoagulant (LA) and high titers of IgG anti-cardiolipin (aCL) are at increased risk [80,81][61][62].
Clinical manifestations depend on the severity of vasculature occlusion, ranging from asymptomatic small vessels occlusion to massive PE with sudden right ventricular failure and acute circulatory collapse. Other symptoms of PE include pleuritic chest pain, dyspnea, hemoptysis, crepitations, tachypnea and tachycardia. Chronic PE can progress to secondary pulmonary arterial hypertension (PAH) due to the reduction of pulmonary vascular tree [49][30]. In addition to PAH, other non-thrombotic intrathoracic manifestations of APS associated with SLE are: DAH, adult respiratory distress syndrome (ARDS) and valvular heart disease (e.g., Libman-Sacks endocarditis) [49,82][30][63]. A rare, potentially fatal, manifestation of APS is the catastrophic APS (CAPS). CAPS is characterized by the diffuse occlusion of small vessels in three or more organs [81,82,83,84,85][62][63][64][65][66]. It generally develops in APS patients in association with a trigger such as infections, neoplasm or surgery. Respiratory failure is often present and can rapidly progress to acute respiratory distress syndrome (ARDS) [81,82,83,84,85][62][63][64][65][66].
Treatment of APS includes anticoagulation with the vitamin K antagonists (VKA), to maintain an international normalized ratio (INR) range of 2.0 to 3.0, for a definite period in a first provoked episode, indefinitely in recurrent episodes or in patients with a high-risk profile [81,85][62][66]. In patients with recurrent arterial or venous thrombosis, a higher INR range 3.0–4.0 or the addition on low dose aspirin should be considered. Common CV risk factors should be corrected, concurrently. In high-risk anti-phospholipid antibodies (aPL) carriers without history of thrombosis, prophylactic treatment with low dose aspirin can be adopted [81,85][62][66]. Hydroxychloroquine may reduce thrombotic risk both in APS and non-APS SLE patients due to its pleiotropic effects but evidence in this regard is still scarce [78,85][59][66]. Treatment of CAPS includes: elimination of triggers (e.g., infections), combination therapy with heparin, glucocorticoids and plasma exchange or intravenous immunoglobulins. B-cell depletion (e.g., RTX) or complement inhibition (e.g., eculizumab) can be considered in refractory cases. Supportive treatments in the intensive care unit may be necessary [81,85][62][66]. Recent systematic literature reviews and meta-analyses investigating direct oral anticoagulants have recommended against their use in these patients [86,87][67][68].
4.3. Pulmonary Arterial Hypertension
Pulmonary hypertension (PH) is classified into five major categories, according to its clinical characteristics and etiology and pulmonary arterial hypertension (PAH) associated with connective tissue diseases (CTDs) belongs to the first group and it is the second most frequent form after idiopathic PAH [88,89][69][70]. PAH is defined by the presence of an increase in mean pulmonary arterial pressure (mPAP) ≥ 25mmHg at rest (assessed by right heart catheterization (RHC)) with a normal pulmonary capillary wedge pressure (≤15 mmHg) and increased pulmonary vascular resistance (PVR) > 3 wood units (WU) [73][54]. Less frequently, SLE patients can present PH secondary to chronic pulmonary thromboembolism (group 4), mitral stenosis due to Libman-Sacks endocarditis (group 2), pulmonary veno-occlusive disease (group 1), ILD-associated PH (group 3) [88,89,90,91,92][69][70][71][72][73].
According to the REVEAL registry (Registry to Evaluate Early and Long-term Pulmonary Arterial Hypertension disease management), SLE patients display the second highest prevalence of PAH after systemic sclerosis (SSc) [93,94][74][75]. The real prevalence of PAH among SLE patients is unknown. Past studies have reported different results due to the method used for diagnosis (right heart catheterization (RHC) versus transthoracic echocardiography (TTE)) and the cut-off value used for the diagnosis [94][75]. The majority of patients are women with a mean age at PAH diagnosis of about 45 years, and with its prevalence and severity increasing with time from SLE onset. PAH can occasionally be the first manifestation of SLE. Usually, PAH tends to be moderate with systolic PAP of 40–60 mmHg and PVR between 5 and 15 WU [93,94,95][74][75][76]. Some possible risk factors for PAH are Raynaud’s phenomenon, active renal disease, vasculitic manifestations, pleuritis, pericardial effusion, ILD, SLEDAI ≤9, lack of rash, low erythrocyte sedimentation rate (ESR) ≤ 20 mm/h. Among immunological parameters associated with PAH: aPL, Anti-U1-RNP and anti-SSA/Ro have been described [94][75]. The pathogenesis of SLE-PAH is probably multifactorial and is not completely understood. Multiple factors such as genetic predisposition, environmental stimuli and immune system dysfunction could lead to an imbalance between vasoconstrictor and vasodilator mediators resulting in an increase in PVR [94,96][75][77]. aPL, anti-endothelial cells and anti-endothelin receptor antibodies, vasculitis, vasospasm, inflammation, decreased oxygen saturation, apoptosis and smooth muscle cell proliferation contribute to the development of the typical lesions of idiopathic PAH, such as plexiform lesions, smooth muscle cell hypertrophy, intimal proliferation, and collagen deposition [94,96][75][77]. Moreover, in SLE-associated PAH, there is an involvement of pulmonary veins and perivascular inflammatory infiltration [94,96,97][75][77][78].
5. Pleural Disease
Pleuritis is the most frequent lung manifestation in patients with SLE, occurring, often in association with pericarditis, in about 40–60% of patients during the course of the disease, although in autoptic studies up to 83% of patients can show signs of pleural involvement [10,112][10][79]. Of note, it is the only SLE manifestation of the respiratory system included in the diagnostic criteria [5]. Pleuritis, with or without pleural effusion, can be the first manifestation of SLE in the 3% and 1% of SLE patients, respectively [113,114][80][81]. Pleural involvement can be present also in overlap syndromes like rhupus syndrome [115][82]. The clinical picture can vary from asymptomatic, incidental findings on imaging, to pleuritic chest pain that is increased with deep inspiration, dyspnea, dry cough, fever and other systemic manifestations. Pleural effusion can be uni- or bilateral, usually mild to moderate, rarely massive. Occasionally pleuritis can be dry [10,49][10][30]. Pathogenesis of pleural effusion is thought to be due to ICs deposition on pleural surfaces. Histopathologic studies have shown the presence of a non-specific lymphoplasmacytic infiltration with rare evidence of IC-mediated vasculitis [115][82]. Pleuritic fluid is sterile, exudative, and yellow-tinged, but occasionally it can be turbidous or seroematic. It contains inflammatory cells such as neutrophils, but it can show a predominance of mononuclear lymphocytic cells, especially in longstanding cases. It also contains glucose levels similar to those of plasma (60–95 mg/dL), increased levels of adenosine deaminase, decreased levels of complement and ANA, in particular with titer ≥ 1:160. It has a greater pH (>7.35) and lower lactate dehydrogenase (LDH) levels (<500 IU/L or <2 times upper limit of normal for serum) than in patients with RA or tuberculosis. LE cells can be seen showing a low sensibility (about 40%) and a specificity of 80%. However, none of these characteristics are specific to SLE pleuritis [10,49,113,115,116,117][10][30][80][82][83][84]. Differential diagnosis may be difficult, since SLE patients can have pleural effusions for many reasons including infections, renal and cardiac failure, pulmonary embolism, and rarely malignancies. It is interesting to note that in SLE pleuritis CRP can be elevated also in the absence of infections [116][83]. Pleural biopsy can occasionally be necessary, only to rule out tuberculosis or malignancy [116][83]. Prognosis is usually favorable, with a good and rapid response to CS at medium dosage, although development of progressive pleural fibrosis leading to fibrothorax has been described. Non-steroidal anti-inflammatory drugs (NSAIDs) can be used for milder cases and spontaneous resolution can also occur. In more severe cases CS can be used (in patients already on steroid therapy an increase of dosages may be needed). In chronic forms, hydroxychloroquine can be used as a glucocorticoid-sparing agent. Major immunosuppressants (e.g., CYC and azathioprine) are not used, unless in the case of a concomitant systemic involvement. An association of IVIg and cyclosporine has been used in chronic, refractory pleural effusion. Chest drainage, pleurodesis and/or pleurectomy are rarely necessary in severe refractory cases [51,113,117,118,119][32][80][84][85][86].
References
- Stojan, G.; Petri, M. Epidemiology of Systemic Lupus Erythematosus: An update. Curr. Opin. Rheumatol. 2018, 30, 144–150.
- Zucchi, D.; Elefante, E.; Calabresi, E.; Signorini, V.; Bortoluzzi, A.; Tani, C. One year in review 2019: Systemic lupus erythematosus. Clin. Exp. Rheumatol. 2019, 37, 715–722.
- Cervera, R.; Doria, A.; Amoura, Z.; Khamashta, M.; Schneider, M.; Guillemin, F.; Maurel, F.; Garofano, A.; Roset, M.; Perna, A.; et al. Patterns of systemic lupus erythematosus expression in Europe. Autoimmun. Rev. 2014, 13, 621–629.
- Lisnevskaia, L.; Murphy, G.; Isenberg, D. Systemic lupus erythematosus. Lancet 2014, 384, 1878–1888.
- Aringer, M.; Costenbader, K.; Daikh, D.; Brinks, R.; Mosca, M.; Ramsey-Goldman, R.; Smolen, J.S.; Wofsy, D.; Boumpas, D.T.; Kamen, D.L.; et al. 2019 European League Against Rheumatism/American College of Rheumatology classification criteria for systemic lupus erythematosus. Ann. Rheum. Dis. 2019, 78, 1151–1159.
- Fanouriakis, A.; Kostopoulou, M.; Alunno, A.; Aringer, M.; Bajema, I.; Boletis, J.N.; Cervera, R.; Doria, A.; Gordon, C.; Govoniet, M.; et al. 2019 update of the EULAR recommendations for the management of systemic lupus erythematosus. Ann. Rheum Dis. 2019, 78, 736–745.
- Kostopoulou, M.; Fanouriakis, A.; Cheema, K.; Boletis, J.; Bertsias, G.; Jayne, D.; Boumpas, D.T. Management of lupus nephritis: A systematic literature review informing the 2019 update of the joint EULAR and European Renal Association-European Dialysis and Transplant Association (EULAR/ERA-EDTA) recommendations. RMD Open. 2020, 6, e001263.
- Al-Adhoubi, N.K.; Bystrom, J. Systemic lupus erythematosus and diffuse alveolar hemorrhage, etiology and novel treatment strategies. Lupus 2020, 29, 355–363.
- Pego-Reigosa, J.M.; Medeiros, D.A.; Isenberg, D.A. Respiratory manifestations of systemic lupus erythematosus: Old and new concepts. Best Pract. Res. Clin. Rheumatol. 2009, 23, 469–480.
- Torre, O.; Harari, S. Pleural and pulmonary involvement in systemic lupus erythematosus. Presse Med. 2011, 40 Pt 2, e19–e29.
- Fidler, L.; Keen, K.J.; Touma, Z.; Mittoo, S. Impact of pulmonary disease on patient-reported outcomes and patient-performed functional testing in systemic lupus erythematosus. Lupus 2016, 25, 1004–1011.
- Karim, A.; Ahmed, S.; Siddiqui, R.; Marder, G.S.; Mattana, J. Severe Upper Airway Obstruction from Cricoarytenoiditis as the Sole Presenting Manifestation of a Systemic Lupus Erythematosus Flare. Chest 2002, 121, 990–993.
- Schwartz, I.S.; Grishman, E. Rheumatoid Nodules of the Vocal Cords as the Initial Manifestation of Systemic Lupus Erythematosus. JAMA 1980, 244, 2751–2752.
- Teitel, A.D.; MacKenzie, C.R.; Stern, R.; Paget, S.A. Laryngeal involvement in systemic lupus erythematosus. Semin. Arthritis Rheum. 1992, 22, 203–214.
- Malinvaud, D.; Mukundan, S.; Crevier-Buchman, L.; Bonfils, P.; Laccourreye, O. Glottic Bamboo Nodules from Systemic Lupus Erythematosus. Ann. Otol. Rhinol. Laryngol. 2013, 122, 496–499.
- Carmiera, D.; Marchand-Adam, S.; Diot, P.; Diot, E. Respiratory involvement in systemic lupus Erythematosus. Rev. Mal. Respir. 2010, 27, e66–e78.
- Luo, Y.; Fan, X.; Jiang, C.; Ramos-Rodriguez, A.; Wen, Y.; Zhang, J.; Huang, F.; Guan, X.; Xu, J. Systemic Lupus Erythematosus and Angioedema: A Cross-Sectional Study From the National Inpatient Sample. Arch. Rheumatol. 2019, 34, 301–307.
- Kumar, N.; Surendran, D.; Bammigatti, C. Angioedema as the presenting feature of systemic lupus erythematosus. BMJ Case Rep. 2018, 2018, bcr2018224222.
- Todic, J.; Leuchter, I. Lupus of the larynx: When bamboo nodes lead to diagnosis. BMJ Case Rep. 2018.
- Lee, J.H.; Sung, I.Y.; Park, J.H.; Roh, J.L. Recurrent laryngeal neuropathy in a systemic lupus erythematosus (SLE) patient. Am. J. Phys. Med. Rehabil. 2008, 87, 68–70.
- Gari, A.G.; Telmesani, A.; Alwithenani, R. Pulmonary Manifestations of Systemic Lupus Erythematosus. In Systemic Lupus Erythematosus; InTech: London, UK, 2012.
- Cantero, C.; Vongthilath, R.; Plojoux, J. Acute lupus pneumonitis as the initial presentation of systemic lupus erythematosus. BMJ Case Rep. 2020, 13, e234638.
- Chattopadhyay, B.; Chatterjee, A.; Maiti, A.; Debnath, N.B. Systemic lupus erythematosus presenting as acute lupus pneumonitis in a young female. J. Postgrad. Med. 2015, 61, 129–130.
- Comer, M.; D’Cruz, D.; Thompson, I.; Erskine, K.; Dacre, J. Pneumonitis in a lupus twin pregnancy: A case report. Lupus 1996, 5, 146–148.
- Osler, W. On the visceral manifestations of the erythema group of skin diseases [Third Paper] 1904. Am. J. Med. Sci. 2009, 338, 396–408.
- Lara, A.R.; Schwarz, M.I. Diffuse Alveolar Hemorrhage. Chest 2010, 137, 1164–1171.
- Quartuccio, L.; Bond, M.; Isola, M.; Monti, S.; Felicetti, M.; Furini, F.; Murgia, S.; Berti, A.; Silvestri, E.; Pazzola, G.; et al. Alveolar haemorrhage in ANCA-associated vasculitis: Long-term outcome and mortality predictors. J. Autoimmun. 2020, 108, 102397.
- Andrade, C.; Mendonca, T.; Farinha, F.; Correia, J.; Marinho, A.; Almeida, I.; Vasconcelos, C. Alveolar hemorrhage in systemic lupus erythematosus: A cohort review. Lupus 2016, 25, 75–80.
- Chen, Y.; Wang, Y.; Chen, X.; Liang, H.; Yang, X. Association of Interstitial Lung Disease with Clinical Characteristics of Chinese Patients with Systemic Lupus Erythematosus. Arch. Rheumatol. 2020, 35, 239–246.
- Tselios, K.; Urowitz, M.B. Cardiovascular and Pulmonary Manifestations of Systemic Lupus Erythematosus. Cur. Rheumatol. Rev. 2017, 13, 206–218.
- Alunno, A.; Gerli, R.; Giacomelli, R.; Carubbi, F. Clinical, Epidemiological, and Histopathological Features of Respiratory Involvement in Rheumatoid Arthritis. Biomed. Res. Int. 2017, 2017, 7915340.
- Keane, M.P.; Lynch, J.P., III. Pleuropulmonary manifestations of systemic lupus erythematosus. Thorax 2000, 55, 159–166.
- Toyoda, Y.; Koyama, K.; Kawano, H.; Nishimura, H.; Kagawa, K.; Morizumi, S.; Naito, N.; Sato, S.; Yamashita, Y.; Takahashi, N.; et al. Clinical features of interstitial pneumonia associated with systemic lupus erythematosus. Respir. Investig. 2019, 57, 435–443.
- Lian, F.; Zhou, J.; Wang, Y.; Cui, W.; Chen, D.; Li, H.; Qiu, Q.; Zhan, Z.; Ye, Y.; Liang, L.; et al. Clinical features and independent predictors of interstitial lung disease in systemic lupus erythematosus. Int. J. Clin. Exp. Med. 2016, 9, 4233–4242.
- Hyldgaard, C.; Hilberg, O.; Pedersen, A.B.; Ulrichsen, S.P.; Løkke, A.; Bendstrup, E.; Ellingsen, T. A population-based cohort study of rheumatoid arthritis-associated interstitial lung disease: Comorbidity and mortality. Ann. Rheum. Dis. 2017, 76, 1700–1706.
- Pérez, E.R.F.; Daniels, C.E.; Sauver, J.S.; Hartman, T.E.; Bartholmai, B.J.; Eunhee, S.Y.; Ryu, J.H.; Schroeder, D.R. Incidence, prevalence, and clinical course of idiopathic pulmonary fibrosis: A population-based study. Chest 2010, 137, 129–137.
- Swigris, J.J.; Berry, G.J.; Raffin, T.A.; Kuschner, W.G. Lymphoid Interstitial Pneumonia. A Narrative Review. Chest 2002, 122, 2150–2164.
- Garcia, D.; Young, L. Lymphocytic interstitial pneumonia as a manifestation of SLE and secondary Sjogren’s syndrome. BMJ Case Rep. 2013, 2013, bcr2013009598.
- Min, J.K.; Hong, Y.S.; Park, S.H.; Park, J.H.; Lee, S.H.; Lee, Y.S.; Kim, H.H.; Cho, C.S.; Kim, H.Y. Bronchiolitis obliterans organizing pneumonia as an initial manifestation in patients with systemic lupus erythematosus. J. Rheumatol. 1997, 24, 2254–2257.
- Otsuka, F.; Amano, T.; Hashimoto, N.; Takahashi, M.; Hayakawa, N.; Makino, H.; Ota, Z.; Ogura, T. Bronchiolitis obliterans organizing pneumonia associated with systemic lupus erythematosus with antiphospholipid antibody. Intern. Med. 1996, 35, 341–344.
- Gammon, R.B.; Bridges, T.A.; al-Nezir, H.; Alexander, C.B.; Kennedy, J.I., Jr. Bronchiolitis obliterans organizing pneumonia associated with systemic lupus erythematosus. Chest 1992, 102, 1171–1174.
- Gutta, S.; Das, S.; Kodiatte, T.A.; Vimala, L.V. Organising pneumonia in Rhupus syndrome. BMJ Case Rep. 2019, 12, e232326.
- Al-Ghanem, S.; Al-Jahdali, H.; Bamefleh, H.; Khan, A.N. Bronchiolitis obliterans organizing pneumonia: Pathogenesis, clinical features, imaging and therapy review. Ann. Thorac. Med. 2008, 3, 67–75.
- Terwiela, M.; Gruttersa, J.C.; Moorsela, C.H.M. Clustering of immune-mediated diseases in sarcoidosis. Curr. Opin. Pulm. Med. 2019, 25, 539–553.
- Rajoriya, N.; Wotton, C.J.; Yeates, D.G.R.; Travis, S.P.L.; Goldacre, M.J. Immune-mediated and chronic inflammatory disease in people with sarcoidosis: Disease associations in a large UK database. Postgrad Med. J. 2009, 85, 233–237.
- Papaioannides, D.; Korantzopoulos, P.; Latsi, P.; Orphanidou, D. Systemic lupus erythematosus developing in a patient with pulmonary sarcoidosis. Joint Bone Spine 2004, 71, 442–444.
- Schnabel, A.; Barth, J.; Schubert, F.; Gross, W.L. Pulmonary Sarcoidosis Coexisting with Systemic Lupus Erythematosus. Scand. J. Rheumatol. 1996, 25, 109–111.
- Robles-Perez, A.; Dorca, J.; Castellvi, I.; Nolla, J.M.; Molina-Molina, M.; Narvaez, J. Rituximab effect in severe progressive connective tissue disease-related lung disease: Preliminary data. Rheumatol. Int. 2020, 40, 719–726.
- Schnabel, A.; Reuter, M.; Gross, W.L. Intravenous pulse cyclophosphamide in the treatment of interstitial lung disease due to collagen vascular diseases. Arthritis Rheum. 1998, 41, 1215–1220.
- Okada, M.; Suzuki, K.; Matsumoto, M.; Nakashima, M.; Nakanishi, T.; Takada, K.; Horikoshi, H.; Matsubara, O.; Ohsuzu, F. Intermittent intravenous cyclophosphamide pulse therapy for the treatment of active interstitial lung disease associated with collagen vascular diseases. Mod. Rheumatol. 2007, 17, 131–136.
- Austin, H.A., III; Klippel, J.H.; Balow, J.E.; le Riche, N.G.; Steinberg, A.D.; Plotz, P.H.; Decker, J.L. Therapy of lupus nephritis. Controlled trial of prednisone and cytotoxic drugs. N. Engl. J. Med. 1986, 314, 614–619.
- Saketkoo, L.A.; Espinoza, L.R. Experience of mycophenolate mofetil in 10 patients with autoimmune-related interstitial lung disease demonstrates promising effects. Am. J. Med. Sci. 2009, 337, 329–335.
- Fischer, A.; Brown, K.K.; Du Bois, R.M.; Frankel, S.K.; Cosgrove, G.P.; Fernandez-Perez, E.R.; Huie, T.J.; Krishnamoorthy, M.; Meehan, R.T.; Olson, A.L.; et al. Mycophenolate mofetil improves lung function in connective tissue disease-associated interstitial lung disease. J. Rheumatol. 2013, 40, 640–646.
- Abramson, S.B.; Dobro, J.; Eberle, M.A.; Benton, M.; Reibman, J.; Epstein, H.; Rapoport, D.M.; Belmont, H.M.; Goldring, R.M. Acute reversible hypoxemia in systemic lupus erythematosus. Ann. Intern. Med. 1991, 114, 941–947.
- Belmont, H.M.; Buyon, J.; Giorno, R.; Abramson, S. Up-regulation of endothelial cell adhesion molecules characterizes disease activity in systemic lupus erythematosus. The Shwartzman phenomenon revisited. Arthritis Rheum. 1994, 37, 376–383.
- Calvo-Alen, J.; Toloza, S.M.A.; Fernandez, M.; Bastian, H.M.; Fessler, B.J.; Roseman, J.M.; McGwin, G., Jr.; Vila, L.M.; Reveille, J.D.; Alarcon, G.S.; et al. Systemic Lupus Erythematosus in a Multiethnic US Cohort (LUMINA) XXV. Smoking, Older Age, Disease Activity, Lupus Anticoagulant, and Glucocorticoid Dose as Risk Factors for the Occurrence of Venous Thrombosis in Lupus Patients. Arthritis Rheum. 2005, 52, 2060–2068.
- Avina-Zubieta, J.A.; Vostretsova, K.; De Vera, M.A.; Sayre, E.C.; Choi, H.K. The risk of pulmonary embolism and deep venous thrombosis in systemic lupus erythematosus: A general population-based study. Semin. Arthritis Rheum. 2015, 45, 195–201.
- Ramirez, G.A.; Efthymiou, M.; Isenberg, D.A.; Cohen, H. Under crossfire: Thromboembolic risk in systemic lupus erythematosus. Rheumatology 2019, 58, 940–952.
- You, H.; Zhao, J.; Wang, Q.; Tian, X.; Li, M.; Zeng, X. Characteristics and risk factors of pulmonary embolism in patients with systemic lupus erythematosus: A case control study. Clin. Exp. Rheumatol. 2020, 38, 940–948.
- Wahl, D.G.; Guillemin, F.; de Maistre, F.; Perret, C.; Lecompte, T.; Thibaut, G. Risk for venous thrombosis related to antiphospholipid antibodies in systemic lupus erythematosus: A meta-analysis. Lupus 1997, 6, 467–473.
- Swigris, J.J.; Fischer, A.; Gilles, J.; Meehan, R.T.; Brown, K.K. Pulmonary and Thrombotic Manifestations of Systemic Lupus Erythematosus. Chest 2008, 133, 271–280.
- Tektonidou, M.G.; Andreoli, L.; Limper, M.; Amoura, Z.; Cervera, R.; Costedoat-Chalumeau, N.; Cuadrado, M.J.; Dörner, T.; Ferrer-Oliveras, R.; Hambly, K.; et al. EULAR recommendations for the management of antiphospholipid syndrome in adults. Ann. Rheum. Dis. 2019, 78, 1296–1304.
- Asherson, R.A.; Cervera, R.; Shepshelovich, D.; Shoenfels, Y. Nonthrombotic manifestations of the antiphosphol ipid syndrome: Away from thrombosis? J. Rheumatol. 2006, 33, 1038–1044.
- Asherson, R.A.; Cervera, R.; Piette, J.C.; Shoenfeld, Y.; Espinosa, G.; Petri, M.A.; Lim, E.; Lau, T.C.; Gurjal, A.; Jedryka-Góral, A.; et al. Catastrophic antiphospholipid syndrome: Clues to the pathogenesis from a series of 80 patients. Medicine 2001, 80, 355–377.
- Bucciarelli, S.; Espinosa, G.; Asherson, R.A.; Cervera, R.; Claver, G.; Gómez-Puerta, J.A.; Ramos-Casals, M.; Ingelmo, M. Catastrophic Antiphospholipid Syndrome Registry Project Group, The acute respiratory distress syndrome in catastrophic antiphospholipid syndrome: Analysis of a series of 47 patients. Ann. Rheum. Dis. 2006, 65, 81–86.
- Chaturvedi, S.; McCrae, K.R. Diagnosis and management of the antiphospholipid syndrome. Blood Rev. 2017, 31, 406–417.
- Dufrost, V.; Risse, J.; Zuily, S.; Wahl, D. Direct Oral Anticoagulants Use in Antiphospholipid Syndrome: Are These Drugs an Effective and Safe Alternative to Warfarin? A Systematic Review of the Literature. Curr. Rheumatol. Rep. 2016, 18, 74.
- Sanchez-Redondo, J.; Espinosa, G.; Varillas Delgado, D.; Cervera, R. Recurrent Thrombosis with Direct Oral Anticoagulants in Antiphospholipid Syndrome: A Systematic Literature Review and Meta-analysis. Clin. Ther. 2019, 41, 1839–1862.
- Galiè, N.; Humbert, M.; Vachiery, J.L.; Gibbs, S.; Lang, I.; Torbicki, A.; Simonneau, G.; Peacock, A.; Noordegraaf, A.V.; Beghetti, M.; et al. 2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension. Eur. Heart J. 2016, 37, 67–119.
- Shahane, A. Pulmonary hypertension in rheumatic diseases: Epidemiology and pathogenesis. Rheumatol. Int. 2013, 33, 1655–1667.
- Soler, J.F.; Borg, A.; Mercieca, C. Dyspnoea in lupus. BMJ Case Rep. 2017, 2017, bcr2017220162.
- Kishida, Y.; Kanai, Y.; Kuramochi, S.; Hosoda, Y. Pulmonary venoocclusive disease in a patient with systemic lupus erythematosus. J. Rheumatol. 1993, 20, 2161–2162.
- Aparicio, I.J.; Lee, J.S. Connective tissue disease associated interstitial lung diseases: Unresolved issues. Semin. Respir. Crit. Care Med. 2016, 37, 468–476.
- McGoon, M.D.; Miller, D.P. REVEAL: A contemporary US pulmonary arterial hypertension registry. Eur. Respir. Rev. 2012, 21, 8–18.
- Tselios, K.; Gladman, D.D.; Urowitz, M.B. Systemic lupus erythematosus and pulmonary arterial hypertension: Links, risks, and management Strategies. Open Access Rheumatol. 2016, 9, 1–9.
- Winslow, T.M.; Ossipov, M.A.; Fazio, G.P.; Simonson, J.S.; Redberg, R.F.; Schiller, N.B. Five-year follow-up study of the prevalence and progression of pulmonary hypertension in systemic lupus erythematosus. Am. Heart J. 1995, 129, 510–515.
- Dhala, A. Pulmonary Arterial Hypertension in Systemic Lupus Erythematosus: Current Status and Future Direction. Clin. Dev. Immunol. 2012, 2012, 854941.
- Dorfmüller, P.; Humbert, M.; Perros, F.; Sanchez, O.; Simonneau, G.; Müller, K.M.; Capron, F. Fibrous remodeling of the pulmonary venous system in pulmonary arterial hypertension associated with connective tissue diseases. Hum. Pathol. 2007, 38, 893–902.
- Crestani, B. The respiratory system in connective tissue disorders. Allergy 2005, 60, 715–734.
- So, C.; Imai, R.; Tomishima, Y.; Nishimura, N. Bilateral Pleuritis as the Initial Symptom of Systemic Lupus Erythematosus: A Case Series and Literature Review. Intern. Med. 2019, 58, 1617–1620.
- Dubois, E.L.; Tuffanelli, D.L. Clinical manifestations of systemic lupus erythematosus: Computer analysis of 520 cases. JAMA 1964, 190, 104–111.
- Saha, K.; Saha, A.; Mitra, M.; Panchadhyayee, P. Bilateral pleural effusion with APLA positivity in a case of rhupus syndrome. Lung India 2014, 31, 390–393.
- Choi, B.Y.; Yoon, M.J.; Shin, K.; Lee, Y.J.; Song, Y.W. Characteristics of pleural effusions in systemic lupus erythematosus: Differential diagnosis of lupus pleuritis. Lupus 2015, 24, 321–326.
- Sharma, S.; Smith, R.; Al-Hameed, F. Fibrothorax and severe lung restriction secondary to lupus pleuritis and its successful treatment by pleurectomy. Can. Respir. J. 2002, 9, 335–337.
- Kao, A.H.; Manzi, S. How to manage patients with cardiopulmonary disease? Best Pract. Res. Clin. Rheumatol. 2002, 16, 211–227.
- Sherer, Y.; Langevitz, P.; Levy, Y.; Fabrizzi, F.; Shoenfeld, Y. Treatment of chronic bilateral pleural effusions with intravenous immunoglobulin and cyclosporine. Lupus 1999, 8, 324–327.