Epigenetic regulation and modification govern the transcriptional mechanisms that promote disease initiation and progression, but can also control the oncogenic processes, cell signaling networks, immunogenicity, and immune cells involved in anti-inflammatory and anti-tumor responses.
- epigenetic
- cell signaling
- DNA methylation
- histone methylation
- protein methylation
- methyltransferase
- activator protein 1 (AP-1)
1. Introduction
Activator protein 1 (AP-1) comprises various transcription factor complexes involved in various cellular and physiological responses. It is recognized as a prime combination of extracellular signals, by which cells adapt to changes in their environment [1,2][1][2]. Activation of AP-1 has been linked to the cause of various severe diseases, which include fibrosis, organ injury, and various inflammatory disorders like rheumatoid arthritis, asthma, and psoriasis by increasing transcription of inflammatory and cellular damaging genes such as cytokines, chemokines, inflammatory mediators, and matrix metalloproteinases [3,4,5,6,7][3][4][5][6][7]. In addition, AP-1 activation also results in cancer progression, which is often dysregulated and contributes to tumor progression, disease aggressiveness, and resistance to drug treatment by transcriptional elevation of oncogenic proteins involved in the regulation of cell-cycle, apoptosis, survival, migration, infiltration, invasion, and proliferation of cells (Table 1) [8,9,10,11][8][9][10][11]. In contrast, AP-1 components can act as tumor suppressors that affect upstream oncogenic events such as the MAPK pathway activity [12]. The disruption of AP-1 can trigger different constituents, which stabilize the functional activation of AP-1 proteins. To curtail the role of AP-1 in various pathologies, targeting AP-1 has been proven to be an attractive therapeutic strategy [13].
Table 1. Representative genes expressed by AP-1.
The regulation of AP-1 activity results in post-translational modifications (PTMs) like the phosphorylation of Tyr, Ser, or Thr and the methylation of Lys or Arg residues [18]. In comparison to protein phosphorylation, methylation is a relatively new area of research. The knowledge of AP-1 regulation through methylation is still incomplete. Thus, in this review, we summarize the putative findings on how methyltransferases and protein substrates interact and regulate AP-1 signal transduction differentially, with a focus on how these epigenetic patterns influence the severity and heterogenicity of diseases. Moreover, we explore how we can implement these mechanisms in the development of therapeutics.
Activator protein 1 (AP-1) protein dimers consist of multigene families of proteins and transcriptional factors consisting of the Fos proteins (c-Fos, v-Fos, Fos B, Fra-1, and Fra-2); Jun proteins (c-Jun, v- Jun, Jun B, and Jun D); activating transcription factors (ATF1-4, ATF6, B-ATF, and ATFx); and musculoaponeurotic fibrosarcoma (Maf) proteins (c-Maf, Maf B, Maf G/F/K, and Nrl) [19,20,21,22][19][20][21][22]. These proteins contain conserved basic region leucine zippers (bZIPs) that are responsible for AP-1 dimerization and DNA binding. AP-1 subunits form homodimers or heterodimers for activation and recognize the consensus AP-1 sites TGAG/CTCA, also known as phorbol 12-O-tetradecanoate-13-acetate (TPA), or TPA-responsive elements [21]. AP-1 is an important transcription factor that modulates diverse cellular processes, including cell survival, differentiation, apoptosis, and development. AP-1 is activated by stimuli, including cytokines, chemokines, stress signals, hormones, and oncogenic stimuli. AP-1 activity is modulated at both the transcriptional and post-translational levels [21,23][21][23]. In post-translational regulation, the phosphorylation of AP-1 subunits activates transcriptional abilities that are preferentially mediated by mitogen-associated protein kinases (MAPKs) [23,24[23][24][25],25], since there are numerous reports that MAPKs or their upstream molecules control AP-1 activation [24,26,27,28][24][26][27][28].
Methylation is the addition of a methyl group (-CH3) to a substrate and is mediated by methyltransferases to regulate biochemical responses [29]. The methyl group is donated by S-adenosylmethionine (SAM, also known as AdoMet), which is converted into S-adenosylhomocysteine (SAH, also known as AdoHcy) (Figure 1) [30,31][30][31]. Methylation occurs on DNA, RNA, and proteins (Figure 1). DNA methylation is a powerful key regulator in epigenetic gene transcription. The DNA methyltransferase (DNMT) family, including DNMT1, DNMT2, DNMT3A, DNMT3B, and DNMT3L, catalyzes a process in which the carbon atoms of the cytosine bases of cytosine-guanine pairs, often called CpG-islands (chromosomal locations rich in cytosine-guanine and p, a phosphate group between DNA bases) [32,33,34][32][33][34]. Of the three DNMTs, DNMT1 is associated with the epigenetic recovery of tissue [35]. Protein methylation is a post-translational modification and can occur on arginine (R), lysine (K), histidine (H), and carboxyl groups [36,37][36][37]. Members of the histone family, including H2A, H2B, H3, and H4, are well-known methylated proteins and are generally methylated on lysine and arginine residues. Methylated histones can change chromatin structure, thereby modulating gene expression [29,36][29][36]. Non-histone methylated proteins have also been reported to regulate cellular processes. Protein lysine methyltransferases (PKMTs) and protein arginine methyltransferases (PRMTs) are representative methyltransferase families. PKMTs generate three types of methylated lysine: monomethyl, dimethyl, and trimethyl lysine [38]. In comparison, three different forms of methylated arginine are generated by PRMTs: monomethyl arginine, asymmetric dimethyl arginine, and symmetric dimethyl arginine [39]. While it is well-known that AP-1 is functionally active in various physiological and pathophysiological conditions, the exact mechanisms that control the AP-1 activation pathway in terms of the methylation reaction of AP-1 and its activation pathway still remain unclear.
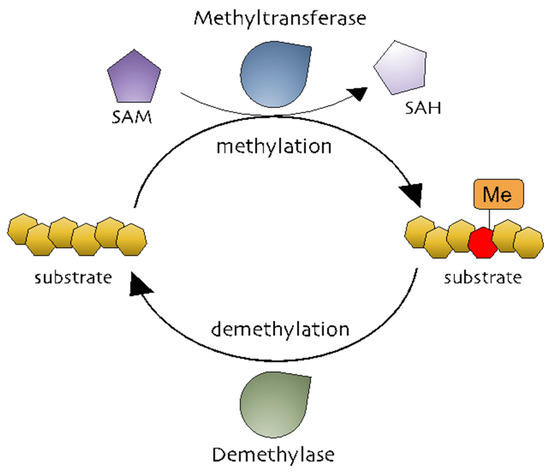
Figure 1. The methylation processes. Methyltransferases transfer a methyl group from a methyl donor (S-adenosylmethionine [SAM]) to a substrate. Methylated substrates can be demethylated by demethylases.
2. DNA Methylation and AP-1 Signaling
2.1. DNA Methylation and Cancer
Accumulating evidence and extensive research have proven that aberrant DNA methylation to be an important epigenetic modulation in activating transcription factors and results in inflammation or different diseases [40]. Small molecule inhibitors of DNA methyltransferase (DNMT), also called as hypomethylating agents, are largely used in therapies for the treatment of myeloid splastic syndrome (MDS), myeloid leukemia. 5-Azacytidine (5-Aza), 5-aza-2’deoxycytidine (decitabine) and SGI-110 (guadecitabine) are DNMT proteins leading to DNA hypomethylation [41,42,43][41][42][43].
Inhibiting or blocking ERK-MAPK signaling significantly reduced DNMT1 protein and gene expression in the SW116 colon cancer cell line, ERK-MAPK inhibitor rottlerin (20 µM) resulted in p16INK4A and p21WAF1 demethylation, which provides a direct link between ERK-MAPK signaling and DNA methylation [44]. Furthermore, a recent investigation showed ROS in the regulation of CDH-1 and E-cadherin plays an important role in the development and progression of breast cancer, by using H2O2 (40 µM) authors have shown induction of cellular migration, DNMT1, HDAC, Snail and Slug (downstream of ERK pathway) and decrease in E-cadherin gene expression and enrichment of H3K9me3 and H3K27me3 in CHD-1 promoter in MDA-MB231 and MCF-7 breast cancer cell lines, treatment of U0126 (ERK inhibitor) reduced gene expression of DNMT-1, Snail and Snug with an increase in E-cadherin and CDH-1, which implies that CDH-1 is symbiotically modulated DNA, histone methylation with histone deacetylation and results in chromatin remodeling and activation of Snail and Snug through the ERK pathway [45]. Hypermethylation of tumor suppressor genes have been related to smoking; exposure to nicotine (10 nM and 10 µM) in human pancreatic epithelial cells resulted in up-regulation of DNMT3A and 3B protein expression with activation of the acetylcholine receptor (a7nACHR) and ERK1/2, JNK and p38 MAPK, combination of ERK1/2 (U0126) and p38 (SB203580) resulted in downregulation of DNMT3A and 3B [46].
Gliomas are the most common forms of brain tumor with poor clinical results and a lower survival rate [47] Relationship between c-Jun, DNMT-1 and global methylation was studied in higher and lower grade gliomas, DNMT-1 mas, with cogene expression being 4.57-fold higher in low grade gliomas in comparison to high grade gliorrelated with overall CpG methylation levels; TCGA (tumor cancer genome atlas analysis ) analysis found that DNMT-1 was also associated with better survival in low grade gliomas with high phosphorylation of c-Jun and high CpG methylation in low grade gliomas. Patient-derived glioblastoma (BTSC168) cells treated with anisomycin increased phosphorylation of JNK, c-Jun and DNMT-1 expression levels and a significant increase in genome wide DNA methylation of promotor regions, whereas JNK inhibitor (SP600125, 50 µmol/mL) reduced protein levels of c-Jun, JNK and DNMT-1 with a reduction in global DNA methylation in CL3021 cells. The results concluded that phospho-c-Jun controls DNMT-1 expression and regulates DNA methylation in glioblastoma [48]. Studies on nasopharyngeal carcinoma (NPC) showed LMP1 induced DNMT-1 protein and RNA expression NP69 (stably expressed LMP1), in addition to siRNA targeting of JNK, c-Jun and TRADD, LMP1-YYD domain and LMP1 observed reduced DNMT-1 expression by 20%, 40%, 60% and 50%. NPC biopsy from 32 patients showed high c-Jun and DNMT-1 and LMP-1 protein expression in 27 of 32 patients (84.38%), which suggests a significant correlation between LPP1-C-Jun-DNMT1 proteins [49].
Subsequent studies have examined the increase of CD38 cell surface expression in multiple myeloma (MM) and could improve daratumumab efficacy and cell resistance. A recent study found that DNA methylation represses CD38 (CpG island in first exon) expression based on ENCODE data. MM cell lines RPMI-8226, MM.1S, XG-1 and KMS12-PE with an increasing dose of azacytidine (AZA) (1–3 µM) resulted in 1.2–2.4-fold increase in CD38 MFI in all the MM cell lines (Table 2). Furthermore ChIP-seq data resulted in transcription factor PU.1 and ATF2 involved in the regulation of CD38 expression; however, knockdown of these genes did not alter AZA-induced CD38 expression and resulted in an increase in TNF-α expression. Cotreatment of AZA with a TNF-α neutralizing antibody completely abrogated CD38 expression in MM plasma cells, suggesting that the TNF-α pathway may play an important role in orchestrating this process [50].
Table 2. List of DNMT1 compounds and their roles.
Compound | Target | Disease | ||||||
|---|---|---|---|---|---|---|---|---|
Azacytidine (5-Aza) | DNMT | Myeloid splastic syndrome Multiple myeloma | ||||||
Decitabine (5-aza-2’-deoxycytidine) | DNMT | Myeloid splastic syndrome | ||||||
Guadecitabine (SGI-110) | DNMT | Myeloid splastic syndrome |
It is shown that CpG methylation in mammalian DNA is known to increase the binding of c-Jun/c-Fos heterodimer [51]. A recent technique involving the use of microfluidic-based ligand enrichment followed by Smile-seq showed a significant difference between DNA binding motifs Jun homodimers and heterodimers [51]. Jun-Fos heterodimers strongly binds to the TPA-response element (TRE). In another report, the analysis of CpG DNA methylation showed that c-Fos/c-Jun heterodimers bind more strongly to mCGACTCA than unmethylated CGACTCA [52].
2.2. DNA Methylation and Osteoporosis
Profound loss of bone leads to changes in skeletal architecture and integrity, and results in disuse osteoporosis (DOP). A study revealed that DNMT-1 levels were significantly higher in hindlimb unloading (HLU) rats with DOP with decreased expression levels of H19 after 3 and 4 weeks with inhibited ERK signaling pathways in DOP bone tissues. The in vivo knockdown of DNMT-1 using SiRNA in Sprague-Dawley (SD) rats significantly upregulated H19 expression with a decrease in CpG methylation rates of 37.0% in the siDNMT group and activation of MAPK-ERK, which prevented the development of DOP; these results suggest that targeting DNMT-1-H19-ERK signaling can be used as a new strategy for treating DOP [53]. A similar work by Lorenzo et al. examined DNA methylation patterns during osteoclastogenesis (OC) [54]. Analysis of TRANSFAC data during OC differentiation revealed the hypomethylation of transcription factors like AP-1 (39%) and NF-κB (15%) with greater enrichment in PU.1 PU.1 is critically involved in OC differentiation and the regulation of cytokine expression and is located upstream of NF-κB. The knockdown of PU.1 in monocytes using siRNA resulted in impaired DNA methylation and reduced TET2 and DNMT3b during OC differentiation.
2.3. DNA Methylation and Inflammation
DNA methylation is also known to regulate TLRs, TNF-receptor associated factor 6 (TRAF6), and myeloid differentiation primary response 88 (MyD88) adaptor proteins, which suggests that DNA methylation can epigenetically regulate inflammatory signaling [44]. A recent report on dental pulp inflammation in human dental pulp cells (hDPCs) challenged with lipopolysaccharide (LPS) decreased DNMT1, IL-6, and IL-8 mRNA gene expression within 24 hours; furthermore, the knockdown of DNMT1 by siRNA increased IL-6 and IL-8 gene expression with activation of the NF-κB and MAPK signaling pathways, suggesting that DNA methylation plays an important role in inflammatory responses [55] (Figure 2). Similar studies on dental caries, a chronic, infection, and destructive disease, observed a decrease in DNMT-1 with a decrease in the gene expression levels of inflammatory cytokines, whereas the knockdown of DNMT-1 resulted in increases in p38 and ERK in the MAPK pathway and resulted in the hypermethylation of the MyD88 adaptor protein in lipoteichoic acid (LTA)-stimulated human odontoblast-like cells (hoBs) [56]. The above studies demonstrate that DNMT-1 is an important regulator of epigenetic modification in controlling inflammation.
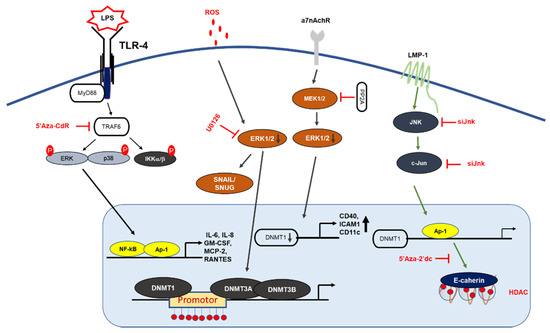
MAPK signaling mechanism by DNA methylation. LMP1 induction of DNMT1 results in hypermethylation of E-cadherin promoter: LMP1 activates the signaling of AP-1, which then binds to the DNMT-1 promoter, leading to hypermethylation of the E-cadherin gene. Deletion of the DNMT1 component increases LPS-induced cytokine expression in NF-κB and AP-1 signaling. PP2A dephosphorylates MEK1/2 and inhibits the activity of DNMT1, leading to the hypomethylation and increased expression of CD70 and CD11a. In T-cells, 5-Aza-Cdr treatment significantly increased the gene expression levels of cytokine IL-6, IL-8 GM-CSF, MCP2, and RANTES, and resulted in the upregulation of ERK phosphorylation.
Human immunodeficiency virus type-1 (HIV-1) infection results in changes in gene expression patterns and the induction of transcription factors NF-κB and AP-1, which regulate DNMT-1 expression. A study on HIV-1 by Youngblood and Reich [57] showed that DNMT-1 luciferase activity was increased by 6- to 7-fold in co-transfected HIV-1 cDNA; blocking AP-1 activity using resveratrol (15 µM) reduced the HIV-1 induction of pGL3DNMT1-SV40 (hybrid construct), so this study suggested that the inhibition of DNMT-1 could reduce HIV-1 pathogenesis [57].
2.4. DNA Methylation and Autoimmune Disease
Systemic lupus erythematous (SLE) is an autoimmune disorder in females characterized by the production of autoantibodies and results in epigenetic changes in DNA and histone [58]. Procainamide, a DNMT inhibitor inhibits ERK pathways by downregulating DNMT expression in lupus T cells [59] (Table 2). Hydralazine suppressed the upregulation of DNMT1 and DNMT3a activity through the Ras/MAPK signaling pathway without inhibiting DNMT activity. Inhibiting ERK and DNA methylation using an inhibitor decreased DNA methyltransferase in lupus T cells, suggesting that DNA methylation and ERK inhibitors may be relevant in lupus recovery and may contribute to the development of autoimmunity [60]. A recent study showed that the silencing of protein phosphatase 2A (PP2Ac) in T-cells using siRNA resulted in increased DNMT1 expression and MEK/ERK phosphorylation with reduced expression of CD70 and ITGAL (methylation-sensitive genes). T-cells isolated from SLE patients also resulted in similar patterns; these reports suggest a potential link between pp2Ac and DNMT1 [61]. A lupus-inducing drug, hydralazine, is reported to contribute to lupus disease pathogenesis [62]. Hydralazine inhibited the ERK pathway, which resulted in the hypomethylation of DNMT1 and DNMT3a in T cells [62]. The results suggest that the inhibition of MAPK/ERK signaling is important for DNA methylation in T cells.
References
- Lopez-Bergami, P.; Lau, E.; Ronai, Z. Emerging roles of ATF2 and the dynamic AP1 network in cancer. Nat. Rev. Cancer 2010, 10, 65–76.
- Hess, J.; Angel, P.; Schorpp-Kistner, M. AP-1 subunits: Quarrel and harmony among siblings. J. Cell Sci. 2004, 117, 5965–5973.
- Zenz, R.; Eferl, R.; Scheinecker, C.; Redlich, K.; Smolen, J.; Schonthaler, H.B.; Kenner, L.; Tschachler, E.; Wagner, E.F. Activator protein 1 (Fos/Jun) functions in inflammatory bone and skin disease. Arthritis Res. Ther. 2007, 10.
- Gungl, A.; Biasin, V.; Wilhelm, J.; Olschewski, A.; Kwapiszewska, G.; Marsh, L.M. Fra2 Overexpression in mice leads to non-allergic asthma development in an IL-13 dependent manner. Front. Immunol. 2018, 9, 2018.
- Trop-Steinberg, S.; Azar, Y. AP-1 expression and its clinical relevance in immune disorders and cancer. Am. J. Med. Sci. 2017, 353, 474–483.
- Yang, W.S.; Kim, J.H.; Jeong, D.; Hong, Y.H.; Park, S.H.; Yang, Y.; Jang, Y.-J.; Kim, J.-H.; Cho, J.Y. 3-Deazaadenosine, an S-adenosylhomocysteine hydrolase inhibitor, attenuates lipopolysaccharide-induced inflammatory responses via inhibition of AP-1 and NF-κB signaling. Biochem. Pharmacol. 2020, 182, 114264.
- Yang, W.S.; Kim, H.G.; Lee, Y.; Yoon, K.; Kim, S.; Kim, J.H.; Cho, J.Y. Isoprenylcysteine carboxyl methyltransferase inhibitors exerts anti-inflammatory activity. Biochem. Pharmacol. 2020, 182, 114219.
- Belguise, K.; Cherradi, S.; Sarr, A.; Boissière, F.; Boulle, N.; Simony-Lafontaine, J.; Choesmel-Cadamuro, V.; Wang, X.; Chalbos, D. PKCθ-induced phosphorylations control the ability of Fra-1 to stimulate gene expression and cancer cell migration. Cancer Lett. 2017, 385, 97–107.
- Talotta, F.; Mega, T.; Bossis, G.; Casalino, L.; Basbous, J.; Jariel-Encontre, I.; Piechaczyk, M.; Verde, P. Heterodimerization with Fra-1 cooperates with the ERK pathway to stabilize c-Jun in response to the RAS oncoprotein. Oncogene 2010, 29, 4732–4740.
- Choi, E.; Kim, E.; Kim, J.H.; Yoon, K.; Kim, S.; Lee, J.; Cho, J.Y. AKT1-targeted proapoptotic activity of compound K in human breast cancer cells. J. Ginseng Res. 2019, 43, 692–698.
- Ahuja, A.; Kim, J.H.; Kim, J.-H.; Yi, Y.-S.; Cho, J.Y. Functional role of ginseng-derived compounds in cancer. J. Ginseng Res. 2018, 42, 248–254.
- Eferl, R.; Wagner, E.F. AP-1: A double-edged sword in tumorigenesis. Nat. Rev. Cancer 2003, 3, 859–868.
- Ye, N.; Ding, Y.; Wild, C.; Shen, Q.; Zhou, J. Small molecule inhibitors targeting activator protein 1 (AP-1). J. Med. Chem. 2014, 57, 6930–6948.
- Wang, A.; Al-Kuhlani, M.; Johnston, S.C.; Ojcius, D.M.; Chou, J.; Dean, D. Transcription factor complex AP-1 mediates inflammation initiated by Chlamydia pneumoniae infection. Cell. Microbiol. 2013, 15, 779–794.
- Ozanne, B.W.; Spence, H.J.; McGarry, L.C.; Hennigan, R.F. Transcription factors control invasion: AP-1 the first among equals. Oncogene 2006, 26, 1–10.
- Benbow, U.; Brinckerhoff, C.E. The AP-1 site and MMP gene regulation: What is all the fuss about? Matrix Biol. 1997, 15, 519–526.
- Shaulian, E.; Karin, M. AP-1 in cell proliferation and survival. Oncogene 2001, 20, 2390–2400.
- Biggar, K.K.; Li, S.S. Non-histone protein methylation as a regulator of cellular signalling and function. Nat. Rev. Mol. Cell Biol. 2015, 16, 5–17.
- Wurm, S.; Zhang, J.; Guinea-Viniegra, J.; García, F.; Muñoz, J.; Bakiri, L.; Ezhkova, E.; Wagner, E.F. Terminal epidermal differen-tiation is regulated by the interaction of Fra-2/AP-1 with Ezh2 and ERK1/2. Genes Dev. 2015, 29, 144–156.
- Shaulian, E.; Karin, M. AP-1 as a regulator of cell life and death. Nat Cell Biol. 2002, 4, E131–E136.
- Karin, M.; Liu, Z.-G.; Zandi, E. AP-1 function and regulation. Curr. Opin. Cell Biol. 1997, 9, 240–246.
- Gazon, H.; Barbeau, B.; Mesnard, J.-M.; Peloponese Jr, J.-M. Hijacking of the AP-1 Signaling pathway during development of ATL. Front. Microbiol. 2018, 8, 2686.
- Karin, M.; Marshall, C.J. The regulation of AP-1 activity by mitogen-activated protein kinases. Phil. Trans. R. Soc. Lond. B 1996, 351, 127–134.
- Tewari, D.; Nabavi, S.F.; Nabavi, S.M.; Sureda, A.; Farooqi, A.A.; Atanasov, A.G.; Vacca, R.A.; Sethi, G.; Bishayee, A. Targeting activator protein 1 signaling pathway by bioactive natural agents: Possible therapeutic strategy for cancer prevention and intervention. Pharmacol. Res. 2017, 128, 366–375.
- Whitmarsh, A.J. Regulation of gene transcription by mitogen-activated protein kinase signaling pathways. Biochim. Biophys. Acta Mol. Cell Res. 2007, 1773, 1285–1298.
- Agron, M.; Brekhman, V.; Morgenstern, D.; Lotan, T. Regulation of AP-1 by MAPK signaling in metal-stressed sea anemone. Cell. Physiol. Biochem. 2017, 42, 952–964.
- Drechsler, Y.; Dolganiuc, A.; Norkina, O.; Romics, L.; Li, W.; Kodys, K.; Bach, F.H.; Mandrekar, P.; Szabo, G. Heme oxygenase-1 mediates the anti-inflammatory effects of acute alcohol on IL-10 induction involving p38 MAPK activation in monocytes. J. Immunol. 2006, 177, 2592–2600.
- Looby, E.; Abdel-Latif, M.M.; Athié-Morales, V.; Duggan, S.; Long, A.; Kelleher, D. Deoxycholate induces COX-2 expression via Erk1/2-, p38-MAPK and AP-1-dependent mechanisms in esophageal cancer cells. BMC Cancer 2009, 9, 1–15.
- Hitchcock, L.N.; Lattal, K.M. Histone-Mediated Epigenetics in Addiction, Progress in Molecular Biology and Translational Science; Elsevier: Amsterdam, The Netherlands, 2014; pp. 51–87.
- Aletta, J.M.; Cimato, T.R.; Ettinger, M.J. Protein methylation: A signal event in post-translational modification. Trends Biochem. Sci. 1998, 23, 89–91.
- Elshorbagy, A.; Jernerén, F.; Samocha-Bonet, D.; Refsum, H.; Heilbronn, L. Serum S-adenosylmethionine, but not methionine, increases in response to overfeeding in humans. Nutr. Diabetes 2016, 6, e192.
- Kim, J.H.; Yoo, B.C.; Yang, W.S.; Kim, E.; Hong, S.; Cho, J.Y. The role of protein arginine methyltransferases in inflammatory responses. Mediat. Inflamm. 2016, 2016.
- Robertson, K.D. DNA methylation, methyltransferases, and cancer. Oncogene 2001, 20, 3139–3155.
- Jin, B.; Li, Y.; Robertson, K.D. DNA methylation: Superior or subordinate in the epigenetic hierarchy? Genes Cancer 2011, 2, 607–617.
- Li, T.; Wang, L.; Du, Y.; Xie, S.; Yang, X.; Lian, F.; Zhou, Z.; Qian, C. Structural and mechanistic insights into UHRF1-mediated DNMT1 activation in the maintenance DNA methylation. Nucleic Acids Res. 2018, 46, 3218–3231.
- Urulangodi, M.; Mohanty, A. DNA damage response and repair pathway modulation by non-histone protein methylation: Implications in neurodegeneration. J. Cell Commun. Signal. 2020, 14, 31–45.
- Lee, D.Y.; Teyssier, C.; Strahl, B.D.; Stallcup, M.R. Role of protein methylation in regulation of transcription. Endocr. Rev. 2005, 26, 147–170.
- Hamamoto, R.; Saloura, V.; Nakamura, Y. Critical roles of non-histone protein lysine methylation in human tumorigenesis. Nat. Rev. Cancer 2015, 15, 110–124.
- Blanc, R.S.; Richard, S. Arginine methylation: The coming of age. Mol. Cell 2017, 65, 8–24.
- Barnicle, A.; Seoighe, C.; Greally, J.M.; Golden, A.; Egan, L.J. Inflammation-associated DNA methylation patterns in epithelium of ulcerative colitis. Epigenetics 2017, 12, 591–606.
- Stresemann, C.; Lyko, F. Modes of action of the DNA methyltransferase inhibitors azacytidine and decitabine. Int. J. Cancer 2008, 123, 8–13.
- Christman, J.K. 5-Azacytidine and 5-aza-2′-deoxycytidine as inhibitors of DNA methylation: Mechanistic studies and their implications for cancer therapy. Oncogene 2002, 21, 5483–5495.
- Juttermann, R.; Li, E.; Jaenisch, R. Toxicity of 5-aza-2’-deoxycytidine to mammalian cells is mediated primarily by covalent trapping of DNA methyltransferase rather than DNA demethylation. Proc. Natl. Acad. Sci. USA 1994, 91, 11797–11801.
- Lu, R.; Wang, X.; Chen, Z.-F.; Sun, D.-F.; Tian, X.-Q.; Fang, J.-Y. Inhibition of the extracellular signal-regulated kinase/mitogen-activated protein kinase pathway decreases dna methylation in colon cancer cells. J. Biol. Chem. 2007, 282, 12249–12259.
- Pradhan, N.; Parbin, S.; Kar, S.; Das, L.; Kirtana, R.; Seshadri, G.S.; Sengupta, D.; Deb, M.; Kausar, C.; Patra, S.K. Epigenetic silencing of genes enhanced by collective role of reactive oxygen species and MAPK signaling downstream ERK/Snail axis: Ectopic application of hydrogen peroxide repress CDH1 gene by enhanced DNA methyltransferase activity in human breast cancer. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 1651–1665.
- Jin, T.; Hao, J.; Fan, D. Nicotine induces aberrant hypermethylation of tumor suppressor genes in pancreatic epithelial ductal cells. Biochem. Biophys. Res. Commun. 2018, 499, 934–940.
- Dunn, G.P.; Rinne, M.L.; Wykosky, J.; Genovese, G.; Quayle, S.N.; Dunn, I.F.; Agarwalla, P.K.; Chheda, M.G.; Campos, B.; Wang, A.; et al. Emerging insights into the molecular and cellular basis of glioblastoma. Genes Dev. 2012, 26, 756–784.
- Heiland, D.H.; Ferrarese, R.; Claus, R.; Dai, F.; Masilamani, A.P.; Kling, E.; Weyerbrock, A.; Kling, T.; Nelander, S.; Carro, M.S. c-Jun-N-terminal phosphorylation regulates DNMT1 expression and genome wide methylation in gliomas. Oncotarget 2016, 8, 6940–6954.
- Tsai, C.-L.; Li, H.-P.; Lu, Y.-J.; Hsueh, C.; Liang, Y.; Chen, C.-L.; Tsao, S.W.; Tse, K.-P.; Yu, J.-S.; Chang, Y.-S. Activation of DNA Methyltransferase 1 by EBV LMP1 Involves c-Jun NH2-Terminal Kinase Signaling. Cancer Res. 2006, 66, 11668–11676.
- Choudhry, P.; Mariano, M.C.; Geng, H.; Martin, T.G.; Wolf, J.L.; Wong, S.W.; Shah, N.; Wiita, A.P. DNA methyltransferase inhibitors upregulate CD38 protein expression and enhance daratumumab efficacy in multiple myeloma. Leukemia 2020, 34, 938–941.
- Isakova, A.; Groux, R.; Imbeault, M.; Rainer, P.; Alpern, D.; Dainese, R.; Ambrosini, G.; Trono, D.; Bucher, P.; Deplancke, B. SMiLE-seq identifies binding motifs of single and dimeric transcription factors. Nat. Methods 2017, 14, 316–322.
- Gustems, M.; Woellmer, A.; Rothbauer, U.; Eck, S.H.; Wieland, T.; Lutter, D.; Hammerschmidt, W. c-Jun/c-Fos heterodimers regulate cellular genes via a newly identified class of methylated DNA sequence motifs. Nucleic Acids Res. 2014, 42, 3059–3072.
- Li, B.; Zhao, J.; Ma, J.-X.; Li, G.-M.; Zhang, Y.; Xing, G.-S.; Liu, J.; Ma, X.-L. Overexpression of DNMT1 leads to hypermethylation of H19 promoter and inhibition of Erk signaling pathway in disuse osteoporosis. Bone 2018, 111, 82–91.
- De La Rica, L.; Rodríguez-Ubreva, J.; García, M.; Islam, A.B.M.M.K.; Urquiza, J.M.; Hernando, H.; Christensen, J.; Helin, K.; Gómez-Vaquero, C.; Ballestar, E. PU.1 target genes undergo Tet2-coupled demethylation and DNMT3b-mediated methylation in monocyte-to-osteoclast differentiation. Genome Biol. 2013, 14, R99.
- Cai, L.; Zhan, M.; Li, Q.; Li, D.; Xu, Q. DNA methyltransferase DNMT1 inhibits lipopolysaccharide-induced inflammatory response in human dental pulp cells involving the methylation changes of IL-6 and TRAF6. Mol. Med. Rep. 2019, 21, 959–968.
- Meng, R.; Li, D.; Feng, Z.; Xu, Q. MyD88 hypermethylation mediated by DNMT1 is associated with LTA-induced inflammatory response in human odontoblast-like cells. Cell Tissue Res. 2019, 376, 413–423.
- Youngblood, B.; Reich, N.O. The early expressed HIV-1 genes regulate DNMT1 expression. Epigenetics 2008, 3, 149–156.
- Zhou, Y.; Lu, Q. DNA methylation in T cells from idiopathic lupus and drug-induced lupus patients. Autoimmun. Rev. 2008, 7, 376–383.
- Scheinbart, L.S.; Johnson, M.A.; Gross, L.A.; Edelstein, S.R.; Richardson, B.C. Procainamide inhibits DNA methyltransferase in a human T cell line. J. Rheumatol. 1991, 18, 530–534.
- Klinman, D.M.; Mushinski, J.F.; Honda, M.; Ishigatsubo, Y.; Mountz, J.D.; Raveche, E.S.; Steinberg, A.D. Oncogene expression in autoimmune and normal peripheral blood mononuclear cells. J. Exp. Med. 1986, 163, 1292–1307.
- Sunahori, K.; Nagpal, K.; Hedrich, C.M.; Mizui, M.; Fitzgerald, L.M.; Tsokos, G.C. The Catalytic subunit of protein phosphatase 2A (PP2Ac) promotes DNA Hypomethylation by suppressing the phosphorylated mitogen-activated protein kinase/Extracellular Signal-regulated Kinase (ERK) Kinase (MEK)/Phosphorylated ERK/DNMT1 Protein pathway in t-cells from controls and systemic lupus erythematosus patients. J. Biol. Chem. 2013, 288, 21936–21944.
- Deng, C.; Lu, Q.; Zhang, Z.; Rao, T.; Attwood, J.; Yung, R.; Richardson, B. Hydralazine may induce autoimmunity by inhibiting extracellular signal-regulated kinase pathway signaling. Arthritis Rheum. 2003, 48, 746–756.