The RBM20 gene encodes the muscle-specific splicing factor RNA-binding motif 20, a regulator of heart-specific alternative splicing. Nearly 40 potentially deleterious variants in RBM20 have been reported in the last ten years, being found to be associated with highly arrhythmogenic events in familial dilated cardiomyopathy. Frequently, malignant arrhythmias can be a primary manifestation of disease. The early recognition of arrhythmic genotypes is crucial in avoiding lethal episodes, as it may have an impact on the adoption of personalized preventive measures.
- RBM20
- Arrhythmias
- Familial dilated cardiomyopathy
1. Introduction
RBM20 gene (Gene ID: 282996; HGNC: 27424; OMIM: 613171; Gencode Gene: ENSG00000203867.7) is located on the long arm of chromosome 10 at position 25.2 (10q25.2) and it encodes the RNA binding motif protein-20 (RBM20). This gene comprises 14 exons (UniProtKB: Q5T481; RefSeq: NM_001134363; Gencode Transcript: ENST00000369519.3) that encode three conserved functional domains: two zinc finger (ZnF) domains and one RNA recognition motif (RRM)-type RNA-binding domain. In addition, sequence alignment from various vertebrate species shows three other conserved regions: a leucine (L)-rich region at the N-terminus, an arginine/serine (RS)-rich region just downstream from the RRM domain, and a glutamate (E)-rich region between the RS-rich region and ZnF2 domain [1] (
Figure 1). The phosphorylation of arginine–serine–arginine–serine–proline residues in the RS region (RSRSP stretch) is necessary for RBM20 nuclear localization [2].
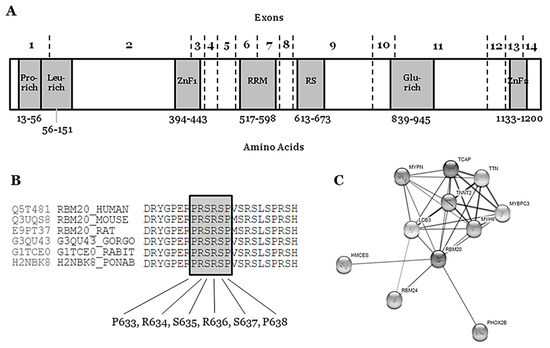
Figure 1.
A
B
C
RBM20 gene is highly expressed during human fetal development (mainly 11–20 weeks of gestation) and in heart and skeletal muscle [3]. The protein (length: 1227 amino acids; mass: 134,357 Da) binds RNA and regulates the splicing of a subset of genes that are involved in cardiac development [4]. It is one of the few heart-specific splicing factors that regulate alternative splicing events of many genes, including
TTN
LDB3 [5][6], and it is associated with sarcomere assembly, ion transport, and diastolic function [7], as well as the expression of calcium handling, rendering high arrhythmic risk to
RBM20 carrier patients [8] (
RBM20
2+
RBM20
RBM20
RBM20
RBM20
RBM20
RBM20
RBM20 mutation p.Ser635Ala shifted human titin isoform expression with an increased molecular weight that was similar to the larger isoform expressed in heterozygous rats [7].
2. Rare RBM20 Variants in Familial Dilated Cardiomyopathy
RBM20 pathogenic variants are at a high risk of AR-DCM and early ICD implantation should be discussed [9]. We performed an exhaustive review of the literature concerning
RBM20
www.ncbi.nlm.nih.gov/clinvar/intro
www.elsevier.com/solutions/embase-biomedical-research
www.ieeexplore.ieee.org/Xplore/home.jsp
www.uniprot.org). The variants were classified according to the American College of Medical Genetics and Genomics/Association for Molecular Pathology (ACMG/AMP) standards and guidelines for the interpretation of sequence variants [10] and described using the HGVS recommendations for the description of sequence variants [11][12]. Concerning frequency of disease-causing variants that are associated with rare inherited diseases, the vast majority of deleterious variants are extremely rare (<0.01%) [13]. ClinGen (
RBM20
Table 1.
RBM20
| Nucleotide Change | Protein Change | dbSNP | gnomAD (MAF%) | ClinVar (Disease) | HGMD (Disease) | CC | ACMG Score | RBM20 Domain | Arrhythmogenic Phenotype |
|---|---|---|---|---|---|---|---|---|---|
| c.247C > A | p.(Leu83Ile) | rs536357058 | 1/155140 (0.0006%) | VUS (DCM) | CM1111132 | VUS | VUS | Exon 2 | Yes |
| (DM; DCM) | Leucine-rich region | ||||||||
| c.680G > T | p.(Gly227Val) | rs202238753 | 225/185204 (0.12%) | LB (DCM) | CM1821953 | LB | VUS | Exon 2 | No |
| (DM; DCM) | |||||||||
| c.769A > G | p.(Thr257Ala) | rs1418674149 | 1/153900 (0.0006%) | NA | CM1815813 | VUS | VUS | Exon 2 | Yes |
| (DM; DCM) | |||||||||
| c.1175G > A | p.(Arg392Gln) | rs751788298 | 3/185862 (0.0016%) | NA | CM1815814 | VUS | VUS | Exon 2 | NA |
| (DM; DCM) | |||||||||
| c.1364C > T | p.(Ser455Leu) | rs189569984 | 862/153884 (0.56%) | LB | NA | LB | LB | Exon 4 | No |
| c.1494C > A | p.(Ser498Arg) | rs774916799 | 2/153882 (0.0013%) | VUS (DCM) | CM1815816 | VUS | VUS | Exon 4 | Yes |
| (DM; DCM) | |||||||||
| c.1528-1G > C | - | rs534513476 | NA | NA | CS183215 | VUS | P | Intron 5–6 | Yes |
| (DM; DCM) | |||||||||
| c.1603G > A | p.(Val535Ile) | rs183007628 | 6/188686 (0.0031%) | VUS (DCM) | CM107458 | VUS | VUS | Exon 6 | Yes |
| (DM; DCM) | RNA Recognition Motif | ||||||||
| c.1760T > A | p.(Leu587His) | NA | NA | NA | CM1815817 | VUS | VUS | Exon 7 | Yes |
| (DM; DCM) | RNA Recognition Motif | ||||||||
| c.1764T > G | p.(Ile588Met) | NA | NA | NA |
gene was reported in 2009 as a novel cause for familial DCM [17]. In this first cross-sectional study, pathogenic variants in
RBM20 were present in the DCM families showing high penetrance, a tent to young age at diagnosis, a notable presence of end-stage heart failure, and high mortality, according to the available information in the included individuals [18]. Nowadays, nearly 30 rare variants in
RBM20 have been reported, explaining 2–3% of DCM [31][32] and supporting an aggressive arrhythmogenic phenotype with a higher risk of SCD [15][19][22][33][34] (
Table 2.
RBM20
| Brauch et al., 2009 ( | n | = 39, DCM) | NC | Li et al., 2010 ( | n | = 16, DCM) | NC | Refaat et al., 2012 ( | n | = 8, DCM) | Wells et al., 2013 ( | n | = 19 carriers) | NC | Van den Hoogenhof et al., 2018 ( | n | = 18, DCM) | Hey et al., 2019 ( | n | = 53, DCM) | Parikh et al., 2019 ( | n | = 74, carriers) | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Age diagnosis | 36 ± 13.2 | 37.6 ± 9 | - | 33.8 ± 11.5 | 42 ± 14 | 37 ± 15 | & | 37 ± 15 | † | |||||||||||||||
| Males | 19 (49%) | 8 (50%) | 4 (50%) | 14 (82%) | 8 (44%) | 31 (58%) | - | |||||||||||||||||
| Follow-up (months) | 60 (12−204) | - | 27.4 ± 15.7 | - | 71 ± 65 | 86 (24−150) | - | |||||||||||||||||
| Mean LVEF | 35.3 ± 11.5 | 29.3 ± 8.6 | - | 48.8 ± 13 | 37 ± 17 | 32 ± 12 | && | 40 ± 17 | ||||||||||||||||
| FH SCD | 39 (100%) | - | - | - | 13 (72%) | - | 22/43 (51%) | †† | ||||||||||||||||
| NSVT | - | 1 (6%) | - | - | 5 (28%) | - | 21/59 (36%) | |||||||||||||||||
| Sustained VT or VF | 9 (23%) | 1 (6%) | 0 | - | 8 (44%) | ¶ | 11 (21%) | &&& | - | |||||||||||||||
| ICD therapy | - | 0 | 1 (12.5%) | - | - | - | 9/32 (28%) | ††† | ||||||||||||||||
| SCD | 3 (7.7%) | 1 (6%) | 0 | - | - | 6 (11.3%) | &&& | 5/60 (8%)-SCA | ††† | |||||||||||||||
| AF | 3 (7.7%) | 2 (12.5%) | 3 (37.5%) * | 6 (33%) | - | 10/58 (17%) | †††† | |||||||||||||||||
| CM183216 | ||||||||||||||||||||||||
| HTx | 4 (mean age 28.5) | 2 (12.5%) | 1 (12.5%) | 1 (5.2%, 17 years old) | + | - | 11 (21%) | &&&& | 5/74 (7%) | |||||||||||||||
| VUS | ||||||||||||||||||||||||
| VUS | ||||||||||||||||||||||||
| Exon 7 | Yes | |||||||||||||||||||||||
| NC | (DM; DCM) | RNA Recognition Motif | ||||||||||||||||||||||
| c.1880 + 4_1880 + 6dupAGG | - | rs1227694990 | 200/187706 (0.1%) | LB (DCM) | CI1516347 | VUS | VUS | Intron 7−8 | No | |||||||||||||||
| Death | 11 (28%, mean age 45): 4 HF (mean age 54.7), 3 SCD (mean age 39) | 3 (11.5%) | 0 | (DM; DCM) | ||||||||||||||||||||
| 11 | (57.9%) | + | - | 2 (4%, end-stage HF at 54 and 73 years old) | 3/74 (4%) | NC | c.1898C > T | p.(Pro633Leu) | rs747880281 | 1/151498 (0.0006%) | VUS (DCM) | NA | VUS | P | Exon 9 | Yes | ||||||||
| Arginine-Serine Domain | ||||||||||||||||||||||||
| c.1900C > T | p.(Arg634Trp) | NA | NA | NA | CM107456 | VUS | LP | Exon 9 | Yes | |||||||||||||||
| (DM; DCM) | Arginine-Serine Domain | |||||||||||||||||||||||
| c.1901G > A | p.(Arg634Gln) | rs267607001 | 1/152378 (0.0006%) | P (DCM) | CM095004 | VUS | LP | Exon 9 | Yes | |||||||||||||||
| c.1901G > T | p.(Arg634Leu) | (DM; DCM) | Arginine-Serine Domain | |||||||||||||||||||||
| c.1903T > G | p.(Ser635Ala) | NA | NA | NA | CM125867 | VUS | LP | Exon 9 | Yes | |||||||||||||||
| (DM; DCM) | Arginine-Serine Domain | |||||||||||||||||||||||
| c.1906C > A | p.(Arg636Ser) | rs267607002 | NA | NA | CM095005 | LP | LP | Exon 9 | Yes | |||||||||||||||
| (DM; DCM) | Arginine-Serine Domain | |||||||||||||||||||||||
| c.1906C > T | p.(Arg636Cys) | rs267607002 | NA | NA | CM107457 | LP | LP | Exon 9 | Yes | |||||||||||||||
| (DM; DCM) | Arginine-Serine Domain | |||||||||||||||||||||||
| c.1907G > A | p.(Arg636His) | rs267607004 | NA | NA | CM095006 | VUS | LP | Exon 9 | Yes | |||||||||||||||
| (DM; DCM) | Arginine-Serine Domain | |||||||||||||||||||||||
| c.1909A > G | p.(Ser637Gly) | rs267607005 | NA | NA | CM095007 | VUS | LP | Exon 9 | Yes | |||||||||||||||
| (DM; DCM) | Arginine-Serine Domain | |||||||||||||||||||||||
| c.1913C > T | p.(Pro638Leu) | rs267607003 | NA | NA | CM095008 | VUS | LP | Exon 9 | Yes | |||||||||||||||
| (DM; DCM) | Arginine-Serine Domain | |||||||||||||||||||||||
| c.1997G > A | p.(Arg666Gln) | rs202011408 | 5/154830 (0.003%) | NA | CM1716804 | VUS | VUS | Exon 9 | Yes | |||||||||||||||
| (DM; DCM) | Arginine-Serine Domain | |||||||||||||||||||||||
| c.2021A > G | p.(Asp674Gly) | rs1475557145 | 1/155286 (0.0006%) | VUS (DCM) | NA | VUS | VUS | Exon 9 | NA | |||||||||||||||
| c.2042A > G | p.(Tyr681Cys) | rs372048968 | 23/186630 (0.01%) | VUS (DCM) | CM1815818 | LB | LB | Exon 9 | No | |||||||||||||||
| (DM; DCM) | ||||||||||||||||||||||||
| c.2062C > T | p.(Arg688Ter) | rs794729150 | 1/31344 (0.003%) | VUS (DCM) | CM1516720 | VUS | P | Exon 9 | Yes | |||||||||||||||
| (DM; DCM) | ||||||||||||||||||||||||
| c.2109G > C | p.(Arg703Ser) | rs988797559 | 2/186026 (0.001%) | NA | CM1111134 | VUS | VUS | Exon 9 | Yes | |||||||||||||||
| (DM; DCM) | ||||||||||||||||||||||||
| c.2147G > A | p.(Arg716Gln) | rs375798246 | 21/155108 (0.013%) | VUS (DCM) | NA | LB | LB | Exon 9 | No | |||||||||||||||
| c.2282G > A | p.(Arg761Gln) | rs556897484 | 4/156496 (0.002%) | NA | NA | VUS | VUS | Exon 9 | NA | |||||||||||||||
| c.2662G > A | p.(Asp888Asn) | rs201370621 | 603/155726 (0.3%) | VUS (DCM) | NA | LB | LB | Exon 11 | No | |||||||||||||||
| c.2737G > A | p.(Glu913Lys) | rs397516607 | NA | LP (DCM) | NA | LP | LP | Exon 11 | Yes | |||||||||||||||
| c.2741T > C | p.(Val914Ala) | rs794729154 | NA | NA | NA | VUS | VUS | Exon 11 | Yes | |||||||||||||||
| c.2714T > A | p.(Met950Lys) | NA | NA | NA | NA | VUS | VUS | Exon 11 | NA | |||||||||||||||
| c.3091G > T | p.(Gly1031Ter) | rs794729157 | NA | NA | CM1111136 | VUS | P | Exon 11 | Yes | |||||||||||||||
| (DM; DCM) | ||||||||||||||||||||||||
| c.3115C > T | p.(Pro1039Ser) | rs727503392 | 40/188260 (0.02%) | LB (DCM) | CM1815819 | VUS | LB | Exon 11 | No | |||||||||||||||
| (DM; DCM) | ||||||||||||||||||||||||
| c.3242C > G | p.(Pro1081Arg) | rs1268330519 | NA | NA | CM1111137 | VUS | VUS | Exon 12 | Yes | |||||||||||||||
| (DM; DCM) | ||||||||||||||||||||||||
| c.3545G > A | p.(Arg1182His) | rs563762318 | 47/185298 (0.025%) | LB (DCM) | CM1510988 | VUS | VUS | Exon 12 | Yes | |||||||||||||||
| (DM; DCM) | Zinc Finger domain 2 | |||||||||||||||||||||||
| c.3616G > A | p.(Glu1206Lys) | rs757389650 | 8/181254 (0.004%) | VUS (DCM) | CM1111138 | VUS | VUS | Exon 14 | NA | |||||||||||||||

Rare variants that are definitely classified as LB can be discarded as causal for FDCM, mainly due to high frequency in the population. However, we cannot discard their potential role as phenotype modifiers. No VUS can be discarded as a potential cause of FDCM—a variant currently classified as VUS means that conclusive data do not exist, so additional studies are needed in order to clarify the definite role in FDCM. These 18 rare non-synonymous VUS in RBM20 should be interpreted with caution by a group of experts, as clinical translation should be personalized, accounting for not only all published data, but also family segregation and phenotype of each patient [14]. The four rare variants classified as P are located in intron 5–6—c.1528-1G>C/IVS5asG>C-1—the end of exon 9—p.(Pro633Leu), p.(Arg688*)—and exon 11—p.(Gly1031*). These variants are considered definitely P due to their extremely low frequency in global population, in silico predictions and functional studies. Most of the variants classified as LP are located in exon 9 (RS domain, amino acids 634–638), suggesting a hot-spot for malignant arrhythmias in FDCM. Actually, a recent study identified two RBM20 regions (exons 9 and 11) with a significant risk for cardiomyopathy, ventricular and atrial arrhythmias, and even SCD [15]. We only identified one rare variant classified as LP and located out of this hot-spot (p.Glu913Lys, Glutamic acid-rich domain). None of these LP variants can be classified as definitely P for FDCM mainly due to a lack of functional data. However, their highly malignant role is supported by low frequencies in global population databases as well as a conserved domain between species.The hot-spot P-R-S-R-S-P between p.(Pro633) and p.(Pro638) contains crucial amino acids for the protein structure and function (Figure 1) (Table 1). In consequence, any amino acid modification inside this zone implies, a priori, a high probability of damaging effect in the protein structure and function. In the first amino acid of this hot-spot, only one rare variant has been recently reported—p.(Pro633Leu). Clinical, genetic, and functional studies confirmed the pathogenic role of this rare variant [16]. Importantly, in this recent study, the authors also suggested that the upregulation of RBM20 may be a viable therapeutic strategy for RBM20-related DCM. In the amino acid 634, two changes have been reported as LP in FDCM [p.(Arg634Trp) and p.(Arg634Gln), CM107456 and CM095004, respectively] [2][17]. In p.(Ser635), only one variation is reported—p.(Ser635Ala), CM125867 [7]. In p.(Arg636), three changes have been published as LP in FDCM—p.(Arg636Ser), p.(Arg636Cys), and p.(Arg636His) [17][18][19][20]. In the last two amino acids, p.(Ser637) and p.(Pro638), a change has been identified in each—p.(Ser637Gly) and p.(Pro638Leu) [21][22][23][24] (Figure 1) (Table 1). The establishment of hiPSC-CMs shows that pathogenic alterations in some of these amino acids may disorganize the sarcomeric complex [8][25][26]. All these variants were identified in families including aggressive arrhythmogenic phenotypes, occasionally in individuals with discrete structural heart alterations, and with a high penetrance of the disease. Despite this fact, we cannot discard that future studies and additional evidence may allow for the identification of other rare variants that are located in different regions of RBM20 and still not associated with arrhythmogenic phenotypes in FDCM. Finally, with regard to CNVs as being potentially responsible for FDCM, to date no structural alteration has been identified in RBM20. Only two CNVs have been associated with FDCM, one located in LMNA [27] and the other in BAG3 [28], explaining <5% of FDCM cases together [29][30].The first pathogenic variant in the RBM
Reefat et al., studied 283 individuals with DCM from the GRADE cohort (that studies the genetic background of patients carrying an ICD), including only non-transplanted patients and with no heart assist device [22]. The patients were screened for
RBM20
RBM20
RBM20
p
RBM20
p
p = 0.047) [22]. This locus have been validated by large GWAS AF studies in general population [35][36]. Haas et al., investigated gene groups in DCM patients to identify genotype-phenotype correlations. In a cohort of 639 patients, the presence of P, LP or VUS
RBM20
p = 0.002) for ICD carrier status in DCM [31]. Van den Hoogenhof et al., compared 18 DCM patients carrying
RBM20
TTN
RBM20
TTN alteration, despite similar LVEF. No differences in non-sustained VT and AF prevalence were detected [34] (
RBM20
n
TTN
LMNA
LMNA
NS
TTN
p
RBM20
p
TTN
p
LMNA
NS
LMNA,
RBM20 [15] (
RBM20
p
p
p
RBM20
RBM20
RBM20
RBM20
p < 0.001). [33] (
RBM20 pathogenic alterations may cause disturbing cardiac contraction and impair cardiac conduction [18]. The mean age at diagnosis is around the forth decade. LVEF is usually impaired, leading to mild to severe DCM, which can lead to heart failure and eventually heart transplantation, which can be required at very young ages. In the published series, no fatal events have been detected in patients without LV structural abnormalities. However, some of these patients have been diagnosed post-mortem, as SCD was the first manifestation of the disease. Similarly to
LMNA
RBM20 carrier’s males are diagnosed at younger ages and suffer major cardiac events before 40 years old when comparing to females (less than 5%) [33]. Lower LVEF justifies the increased level of SCD and heart transplant in males. Analysis of human induced-pluripotent stem cell (hiPSC)-derived cardiomyocytes (hiPSC-CMs) from DCM patients carrying rare variants in
RBM20 have shown that pathogenic alterations in this gene may disorganize the sarcomeric complex [8][25]. The RBM20 hiPSC-CMs were defective in calcium handling machinery with prolonged levels in the cytoplasm and higher spike amplitude [8][25]. Indeed, this fact supports the malignant arrhythmogenic nature of the rare alterations in this gene [10], thereby requiring patients carrying these variants to be more closely followed together with the adoption of personalized preventive measures.
The stratification of SCD risk has been precluded by the use of heterogeneous subsets of patients with idiopathic DCM and by the use of risk models in which predictions are based on static parameters disregarding the disease course. Furthermore, it is important to consider that a large proportion of the
RBM20
RBM20, so accounting that information in future projects will bring clinical value to such work. Of note is that the aggressiveness of the disease has been demonstrated differently according to biological sex, which suggests that a different clinical approach should be applied accordingly. The current approach to personalized risk stratification for DCM is shifting towards a better characterization of the underlying etiology of DCM, in which genetic study has a paramount value. Family screening is mandatory among patients in order to identify asymptomatic DCM affected individuals at risk of SCD, which can be the first symptom of the disease.