The complement system is part of the innate immune response, where it provides immediate protection from infectious agents and it plays a fundamental role in homeostasis. Complement dysregulation occurs in several diseases, where the tightly regulated proteolytic cascade turns offensive. Prominent examples are atypical hemolytic uremic syndrome, paroxysmal nocturnal hemoglobinuria and Alzheimer’s disease. Therapeutic intervention targeting complement activation may allow treatment of such debilitating diseases.
- complement system
- proteolytic cascade
- convertase
1. The Complement System
The complement system is an efficient weapon of innate immunity, which opsonizes the surface of invading organisms and apoptotic host cells for elimination through phagocytosis and cell lysis. In the innate immune system, pattern recognition molecules (PRMs) bind pathogen-associated molecular patterns and damage-associated molecular patterns [3,4][1][2]. Complement activation also elicits an inflammatory response at the site of infection. The complement cascade can be activated through three distinct pathways; the classical pathway (CP), the lectin pathway (LP), and the alternative pathway (AP) (Figure 1). Both the classical and the lectin pathways initiate by activation of giant complexes formed between an oligomeric pattern recognition molecule and a protease complex that cleaves complement component C4 resulting in deposition of the major fragment C4b on the activator (Figure 1). In the CP, C1q is the pattern recognition molecule and circulates in complex with the serine proteases C1r and C1s (C1 in Figure 1). C1q is a hexamer of trimers, and each trimer is formed by chains A, B and C. The C-terminal parts of the three chains fold into structural entities known as the C1q globular domains, while the N-terminal parts of the three chains are joined in a collagen helix. The C1 complex initiates the CP when C1q recognizes antigen bound immunoglobulin G (IgG) and immunoglobulin M (IgM) (Figure 1), the acute phase proteins CRP, pentraxins and anionic phospholipids as phosphatidylserine on apoptotic and necrotic cell surfaces [5,6][3][4]. C1q similarly recognizes LPS in the cell wall of Gram-negative bacteria as well as viral proteins [7][5]. In the brain, C1q binds Aβ and PrP oligomers, playing a role in Alzheimer’s and prion disease progression, respectively [8,9][6][7]. The vast majority of C1q binding patterns are recognized by the C1q globular domain. Specifically, the C1q B and C chains establish direct interactions with the fragment crystallizable (Fc) moiety of IgG and IgM in immune complexes [3,5,10,11,12,13,14][1][3][8][9][10][11][12]. Activated C1s in the C1 complex cleaves C4 to C4b, and a major conformational change in nascent C4b exposes a reactive thioester group, which may react with a nucleophile leading to covalent attachment of C4b to the activator (Figure 1) [15,16][13][14]. The C4b conformation allows binding of the zymogen C2, to form the proconvertase C4b2. Within the proconvertase, C2 is cleaved by C1s resulting in formation of the C3 convertase C4b2a [17,18,19][15][16][17]. In the related LP, the cascade can be initiated by five different PRMs called mannan-binding lectin (MBL), M-, L-, H-ficolins (or ficolin-1 to 3) and CL-LK [20,21,22][18][19][20] upon binding to conserved carbohydrate structures on the activator surface. The LP PRMs circulate in complex with dimers of the MBL-associated serine proteases (MASPs) 1, 2, and 3 [23,24,25][21][22][23]. MASP-1 and MASP-2 are functional homologs of C1r and C1s in the CP, and upon activation of the LP cascade, C4 is cleaved and the same proconvertase and C3 convertase are assembled as in the CP. Since the C4b2a C3 convertase is the endpoint of both pathways, it is referred to as the CP/LP C3 convertase. C4b2a cleaves its substrate C3, which undergoes a conformational change similar to nascent C4b, and exposes the reactive thioester that forms an ester bond with a hydroxyl nucleophile on the surface (Figure 1) [26,27][24][25].
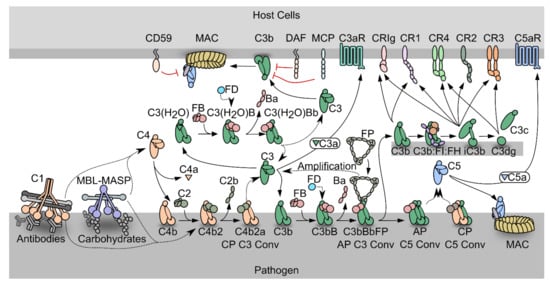
Figure 1. Schematic representation of the complement system. PRM-associated proteases activate upon binding of the PRMs to specific activators. The active proteases cleave C4, which undergoes a conformational change and covalently binds the activator. The zymogen C2 binds the opsonizing C4b, forming the C3 proconvertase. The active C3 convertase cleaves C3, which liberates the C3a fragment from C3b that covalently attaches to the activator. Factor B (FB) may bind the C3b, and upon activation by Factor D, the AP C3 convertase appears, that is stabilized by properdin (FP). C3 may also undergo spontaneous hydrolysis forming C3(H2O), which is a functional homologue of C3b and allows the assembly of a fluid phase C3 convertase. When the C3b concentration reaches a threshold density on the activator, the C3 convertases shifts specificity to C5. The resulting C5 convertases cleave C5 and the C5b forms the starting point for assembly of the membrane attack complex that may perforate the cell membrane. C3b may also undergo degradation by factor I assisted by cofactors. The degradation of C3b opens for interactions with complement receptors.
In the alternative pathway, C3b deposited by the CP/LP C3 convertase binds to factor B (FB) to form the AP proconvertase C3bB (Figure 1). The fluid phase protease Factor D (FD) cleaves FB in the proconvertase and the AP C3 convertase C3bBb appears [28,29,30][26][27][28]. The ability of C3b to form a C3 convertase and promote further C3 cleavage gives rise to the AP amplification loop, which amplifies the C3b deposition catalyzed by the CP/LP C3 convertase by 5-10 fold [31,32,33][29][30][31]. Besides starting from C3b deposited through the CP and LP, a fluid phase AP C3 convertase may assemble after spontaneous C3 hydrolysis (tickover mechanism) whereby a C3b-like molecule called C3(H2O) is formed that is capable of binding to FB [34][32]. This fluid phase proconvertase is also activated by FD cleavage, however the physiological role of the fluid phase AP C3 convertase is still debated [35,36][33][34]. The half-life of the AP C3 convertase is only 90 s under physiological conditions, but it is increased up to ten-fold by binding of properdin (FP), the only known positive regulator of the complement system [37,38][35][36]. FP circulates as dimeric, trimeric, or tetrameric homo-oligomers and recognizes primarily C3b within the AP C3 convertase [39][37].
Both C4b and C3b are tightly regulated to avoid complement activation on host cells, but due to the strong amplification through the alternative pathway, C3b regulation is likely to be the most important in vivo. Regulators of complement activity (RCA) expressed on or recruited to host cells may exert decay acceleration by dissociating the C3 convertases, but may also act as cofactors for C4b and C3b degradation by the protease factor I (FI) [40][38] (Figure 1). The products of FI degradation, C4c C4d, iC3b and C3dg are inactive with respect to convertase formation. The best characterized FI cofactors are factor H (FH), membrane cofactor protein (MCP/CD46) and CR1 (CD35). MCP and CR1 are membrane bound and recognize both C4b and C3b [41,42][39][40]. In contrast, FH is C3b specific and acts both in the fluid phase and on host cells through recognition of sialic acid and glycosaminoglycan [43,44][41][42]. On non-host cell like pathogens, conversion of C3b to iC3b is slower than on host cells due to weaker binding of FH and the lack of membrane bound regulators.
The multiple effector functions elicited by the C3 degradation products are reviewed in [45][43]. C3 cleavage releases the anaphylatoxin C3a, which triggers an inflammatory response at the activation site through binding to the G-protein coupled receptor C3aR [46][44]. Another effector function of C3 fragments is to induce phagocytosis of the activator. C3b and iC3b mediate phagocytosis through interaction with complement receptor Vsig4 (also called CRIg) presented by Kupffer cell macrophages [47[45][46],48], while only iC3b are recognized by CR3 and CR4 to elicit phagocytosis. CR3 and CR4 are expressed in the myeloid subsets of leukocytes, on NK cells and activated T and B lymphocytes [49][47]. Furthermore, iC3b and its degradation product C3dg confer cross talk between the complement cascade and adaptive immunity through their binding to CR2 on B lymphocytes [50][48].
If not degraded by FI, the surface density of C3b continues to increase on the activator. When a threshold density is reached, both C3 convertases shift their substrate specificity from C3 to C5 [51,52][49][50]. C5 is structurally homologous to complement C3 [53][51], but does not have an internal thioester. Instead, C5b interacts with C6, forming the C5b6 complex, which can transiently associate with a nearby lipid bilayer on a complement opsonized cell. Subsequent recruitment of C7 leads to stable association with the membrane, and lipid bilayer penetration starts when C8 joins the complex. The pore size is increased by insertion of several C9 molecules, resulting in formation of the membrane attack complex (MAC) that perforates the cell (Figure 1) [54][52]. The lytic ability of complement is important for killing of Gram negative bacteria, one important example is Neisseria meningitidis [55][53]. The cleavage of C5 also leads to the release of C5a, which triggers a potent inflammatory response through binding to the G-protein coupled receptor C5aR1. This induces vasodilation, release of histamine and contraction of smooth muscle, as well as chemotaxis of neutrophils, T cells, activated B cells, macrophages and basophils [56][54].
2. The Complement System as a Driver of Pathogenesis
Regulators of complement are ubiquitously expressed on host surfaces to control undesired amplification, and tipping of this balance is a major mechanism for diseases associated with complement activation. It is well established that especially aberrant AP activity on self surfaces is associated with the development of atypical hemolytic uremic syndrome (aHUS), paroxysmal nocturnal hemoglobinuria (PNH), age related macular degeneration (AMD), anti-neutrophilic cytoplasmic autoantibodies (ANCA) vasculitis, and C3 glomerulopathy (C3G) [45][43]. PNH arises from clonal expansion of hematopoietic stem cells that comprise a loss-of-function mutation in the PIGA gene [57][55] that is essential for synthesis of glycosylphosphatidylinositol (GPI) anchors and the mutation hence results in deficiency of GPI anchored proteins, including DAF [58][56] and CD59 [59][57]. Lack of these surface bound complement regulators renders erythrocytes vulnerable to complement attack and lysis by the membrane attack complex. The clinical manifestations of the disease include thrombosis and anemia [57][55]. Similarly, complement exacerbates the development of aHUS that arises from dysregulation of complement on host endothelia, most commonly the kidneys [60][58]. Patients suffering from aHUS often possess mutations in FH [61][59] which may result in reduced levels of surface-bound FH and consequently reduced protection against complement activation. Similarly, genetic studies report loss-of-function mutations in genes encoding the complement regulators FI and MCP as well as gain-of-function mutations in genes for FB and C3 in aHUS patients [62][60]. The term ‘C3 glomerulopathy’ is used to describe glomerular disorders, where complement dysregulation either underlies or exacerbates disease development [63][61]. A common characteristic of C3 glomerulopathies (C3G) is C3 fragment deposition in the renal tissue leading to irreversible kidney damage. C3G arise from dysregulation of the AP in fluid phase driven by either acquired or genetic factors. Acquired drivers include autoantibodies, called C3 nephritic factors (C3Nef), that stabilize the C3 convertase [64][62]. In the dense deposit disease subtype of C3G, 78% of patients express C3Nefs [65][63]. Similarly, genetic drivers of C3G commonly lie in the C3 and CFB as well as CFH loci [66][64]. AMD is the leading cause of visual impairment in developed countries. In the early stage of disease development, extracellular deposits of lipids and proteins accumulate between the retinal pigment epithelium and the Bruch’s membrane. Later stages of the disease result in extensive damage of the retinal pigment epithelium and eventually loss of vision [67][65]. An analysis of genetic data from more than 17,000 cases indicated an association between disease and mutations near the genes encoding FH, FI, FB, C2 and C3 [68][66].
The contexts in which the CP of complement is involved are broad, from the homeostatic removal of apoptotic material to induction of heavy inflammation in host tissues. On host cells, tagging by the C1 complex leads to signaling for clearance of the debris. Importantly, this takes place in the absence of inflammation and lysis, due to the action of FI and the regulators, which promptly degrade C3b to iC3b and dissociate the convertases [69][67]. It is well-established that deficiency in the early CP components leads to autoimmunity and development of autoantibodies against neoepitopes on the surface of apoptotic cells, due to impaired clearance of apoptotic material [70][68]. Furthermore, autoimmunity caused by CP activation is also observed in the acute diseases ischemia reperfusion injury, sepsis, antibody induced hemolytic anemia, antibody mediated rejection and cold agglutinin disease [71,72,73,74,75][69][70][71][72][73].
In recent years, it has been established that the classical pathway has a well-defined role in the developmental process of synaptic pruning [76][74], a process required in the developing brain to establish proper synaptic connectivity for a functioning adult brain. Less active synapses are pruned away and one of the mechanisms guiding this process is activation of the complement system through the classical pathway with C4 cleavage, progressing further into the alternative pathway resulting in C3 cleavage [77,78][75][76]. In the neurodevelopmental disorder schizophrenia, genome wide association studies have shown a correlation of a C4 isotype overexpression with disease development, and a polymorphism in the central nervous system (CNS) specific functional homologue of CR1, CSMD1 was identified as a risk factor [79,80,81][77][78][79]. Most recently, C4 overexpression was linked to hypo-connectivity in the prefrontal cortex, and schizophrenia-like symptoms in mice [82][80], and the current hypothesis for schizophrenia pathogenesis involves aberrant complement mediated synaptic pruning. Synaptic pruning onset was also documented during West Nile virus infection and in the neurodegenerative diseases Alzheimer’s, Parkinson’s, multiple sclerosis, frontotemporal dementia and spinal muscular atrophy [83,84,85,86][81][82][83][84]. Hence, the normal function of the CP during development can be aberrantly reactivated with devastating consequences in adulthood [78,83,84,85,87][76][81][82][83][85] and for this reason the CP proteins represent promising therapeutic targets for treatment of neurological diseases with different etiology [86][84].
References
- Bajic, G.; Degn, S.E.; Thiel, S.; Andersen, G.R. Complement activation, regulation, and molecular basis for complement-related diseases. Embo. J. 2015, 34, 2735–2757.
- Janeway, C.A. Approaching the Asymptote? Evolution and Revolution in Immunology. Cold Spring Harb. Symp. Quant. Biol. 1989, 54, 1–13.
- Roumenina, L.T.; Ruseva, M.M.; Zlatarova, A.; Ghai, R.; Kolev, M.; Olova, N.; Gadjeva, M.; Agrawal, A.; Bottazzi, B.; Mantovani, A.; et al. Interaction of C1q with IgG1, C-reactive protein and pentraxin 3: Mutational studies using recombinant globular head modules of human C1q A, B, and C chains. Biochemistry 2006, 45, 4093–4104.
- Païdassi, H.; Tacnet-Delorme, P.; Garlatti, V.; Darnault, C.; Ghebrehiwet, B.; Gaboriaud, C.; Arlaud, G.J.; Frachet, P. C1q Binds Phosphatidylserine and Likely Acts as a Multiligand-Bridging Molecule in Apoptotic Cell Recognition. J. Immunol. 2008, 180, 2329–2338.
- Gaboriaud, C.; Frachet, P.; Thielens, N.M.; Arlaud, G.J. The human c1q globular domain: Structure and recognition of non-immune self ligands. Front. Immunol. 2011, 2, 92.
- Tacnet-Delorme, P.; Chevallier, S.; Arlaud, G.J. β-Amyloid Fibrils Activate the C1 Complex of Complement Under Physiological Conditions: Evidence for a Binding Site for Aβ on the C1q Globular Regions. J. Immunol. 2001, 167, 6374–6381.
- Erlich, P.; Dumestre-Pérard, C.; Ling, W.L.; Lemaire-Vieille, C.; Schoehn, G.; Arlaud, G.J.; Thielens, N.M.; Gagnon, J.; Cesbron, J.-Y. Complement protein C1q forms a complex with cytotoxic prion protein oligomers. J. Biol. Chem. 2010, 285, 19267–19276.
- Ugurlar, D.; Howes, S.C.; de Kreuk, B.-J.; Koning, R.I.; de Jong, R.N.; Beurskens, F.J.; Schuurman, J.; Koster, A.J.; Sharp, T.H.; Parren, P.W.H.I.; et al. Structures of C1-IgG1 provide insights into how danger pattern recognition activates complement. Science 2018, 359, 794–797.
- Bally, I.; Inforzato, A.; Dalonneau, F.; Stravalaci, M.; Bottazzi, B.; Gaboriaud, C.; Thielens, N.M. Interaction of C1q With Pentraxin 3 and IgM Revisited: Mutational Studies With Recombinant C1q Variants. Front. Immunol. 2019, 10.
- Sharp, T.H.; Boyle, A.L.; Diebolder, C.A.; Kros, A.; Koster, A.J.; Gros, P. Insights into IgM-mediated complement activation based on in situ structures of IgM-C1-C4b. Proc. Natl. Acad. Sci. USA 2019, 116, 11900–11905.
- Zlatarova, A.S.; Rouseva, M.; Roumenina, L.T.; Gadjeva, M.; Kolev, M.; Dobrev, I.; Olova, N.; Ghai, R.; Jensenius, J.C.; Reid, K.B.M.; et al. Existence of Different but Overlapping IgG- and IgM-Binding Sites on the Globular Domain of Human C1q. Biochemistry 2006, 45, 9979–9988.
- Diebolder, C.A.; Beurskens, F.J.; de Jong, R.N.; Koning, R.I.; Strumane, K.; Lindorfer, M.A.; Voorhorst, M.; Ugurlar, D.; Rosati, S.; Heck, A.J.R.; et al. Complement is activated by IgG hexamers assembled at the cell surface. Science 2014, 343, 1260–1263.
- Kidmose, R.T.; Laursen, N.S.; Dobó, J.; Kjaer, T.R.; Sirotkina, S.; Yatime, L.; Sottrup-Jensen, L.; Thiel, S.; Gál, P.; Andersen, G.R. Structural basis for activation of the complement system by component C4 cleavage. Proc. Natl. Acad. Sci. USA 2012, 109, 15425–15430.
- Mortensen, S.; Kidmose, R.T.; Petersen, S.V.; Szilágyi, Á.; Prohászka, Z.; Andersen, G.R. Structural Basis for the Function of Complement Component C4 within the Classical and Lectin Pathways of Complement. J. Immunol. 2015, 194, 5488–5496.
- Sitomer, G.; Stroud, R.M.; Mayer, M.M. Reversible adsorption of C′2 by EAC′4: Role of Mg2+, enumeration of competent SAC′4, two-step nature of C′2a fixation and estimation of its efficiency. Immunochemistry 1966, 3, 57–69.
- Müller-Eberhard, H.J.; Polley, M.J.; Calcott, M.A. Formation and functional significance of a molecular complex derived from the second and the fourth component of human complement. J. Exp. Med. 1967, 125, 359–380.
- Wallis, R.; Dodds, A.W.; Mitchell, D.A.; Sim, R.B.; Reid, K.B.M.; Schwaeble, W.J. Molecular Interactions between MASP-2, C4, and C2 and Their Activation Fragments Leading to Complement Activation via the Lectin Pathway. J. Biol. Chem. 2007, 282, 7844–7851.
- Gadjeva, M.; Thiel, S.; Jensenius, J.C. The mannan-binding-lectin pathway of the innate immune response. Curr. Opin. Immunol. 2001, 13, 74–78.
- Matsushita, M.; Fujita, T. Ficolins and the lectin complement pathway. Immunol. Rev. 2001, 180, 78–85.
- Henriksen, M.L.; Brandt, J.; Andrieu, J.-P.; Nielsen, C.; Jensen, P.H.; Holmskov, U.; Jorgensen, T.J.D.; Palarasah, Y.; Thielens, N.M.; Hansen, S. Heteromeric Complexes of Native Collectin Kidney 1 and Collectin Liver 1 Are Found in the Circulation with MASPs and Activate the Complement System. J. Immunol. 2013, 191, 6117–6127.
- Matsushita, M.; Fujita, T. Activation of the classical complement pathway by mannose-binding protein in association with a novel C1s-like serine protease. J. Exp. Med. 1992, 176, 1497–1502.
- Thiel, S.; Vorup-Jensen, T.; Stover, C.M.; Schwaeble, W.; Laursen, S.B.; Poulsen, K.; Willis, A.C.; Eggleton, P.; Hansen, S.; Holmskov, U.; et al. A second serine protease associated with mannan-binding lectin that activates complement. Nature 1997, 386, 506–510.
- Dahl, M.R.; Thiel, S.; Willis, A.C.; Vorup-Jensen, T.; Christensen, T.; Petersen, S.V.; Jensenius, J.C. Mannan-binding lectin associated serine protease 3 (MASP-3)—A new component of the lectin pathway of complement activation. Immunopharmacology 2000, 49, 79.
- Janssen, B.J.C.; Huizinga, E.G.; Raaijmakers, H.C.A.; Roos, A.; Daha, M.R.; Nilsson-Ekdahl, K.; Nilsson, B.; Gros, P. Structures of complement component C3 provide insights into the function and evolution of immunity. Nature 2005, 437, 505–511.
- Janssen, B.J.C.; Christodoulidou, A.; McCarthy, A.; Lambris, J.D.; Gros, P. Structure of C3b reveals conformational changes that underlie complement activity. Nature 2006, 444, 213–216.
- Pangburn, M.K.; Müller-Eberhard, H.J. The C3 convertase of the alternative pathway of human complement. Enzymic properties of the bimolecular proteinase. Biochem. J. 1986, 235, 723–730.
- Rooijakkers, S.H.M.; Wu, J.; Ruyken, M.; van Domselaar, R.; Planken, K.L.; Tzekou, A.; Ricklin, D.; Lambris, J.D.; Janssen, B.J.C.; van Strijp, J.A.G.; et al. Structural and functional implications of the alternative complement pathway C3 convertase stabilized by a staphylococcal inhibitor. Nat. Immunol. 2009, 10, 721.
- Forneris, F.; Ricklin, D.; Wu, J.; Tzekou, A.; Wallace, R.S.; Lambris, J.D.; Gros, P. Structures of C3b in complex with factors B and D give insight into complement convertase formation. Science 2010, 330, 1816–1820.
- Lachmann, P.J. The Amplification Loop of the Complement Pathways. In Advances in Immunology; Alt, F.W., Ed.; Academic Press: Cambridge, MA, USA, 2009; Chapter 4; Volume 104, pp. 115–149.
- Harboe, M.; Garred, P.; Karlstrom, E.; Lindstad, J.K.; Stahl, G.L.; Mollnes, T.E. The down-stream effects of mannan-induced lectin complement pathway activation depend quantitatively on alternative pathway amplification. Mol. Immunol. 2009, 47, 373–380.
- Harboe, M.; Ulvund, G.; Vien, L.; Fung, M.; Mollnes, T.E. The quantitative role of alternative pathway amplification in classical pathway induced terminal complement activation. Clin. Exp. Immunol. 2004, 138, 439–446.
- Fishelson, Z.; Pangburn, M.K.; Müller-Eberhard, H.J. Characterization of the initial C3 convertase of the alternative pathway of human complement. J. Immunol. 1984, 132, 1430–1434.
- Harrison, R.A. The properdin pathway: An "alternative activation pathway" or a "critical amplification loop" for C3 and C5 activation? Semin. Immunopathol. 2018, 40, 15–35.
- Ekdahl, K.N.; Mohlin, C.; Adler, A.; Åman, A.; Anand Manivel, V.; Sandholm, K.; Huber-Lang, M.; Fromell, K.; Nilsson, B. Is generation of C3(H2O) necessary for activation of the alternative pathway in real life? Mol. Immunol. 2019, 114, 353–361.
- Fearon, D.T.; Austen, K.F. Properdin: Binding to C3b and stabilization of the C3b-dependent C3 convertase. J. Exp. Med. 1975, 142, 856–863.
- Hourcade, D.E. The Role of Properdin in the Assembly of the Alternative Pathway C3 Convertases of Complement. J. Biol. Chem. 2006, 281, 2128–2132.
- Pedersen, D.V.; Gadeberg, T.A.F.; Thomas, C.; Wang, Y.; Joram, N.; Jensen, R.K.; Mazarakis, S.M.M.; Revel, M.; El Sissy, C.; Petersen, S.V.; et al. Structural Basis for Properdin Oligomerization and Convertase Stimulation in the Human Complement System. Front. Immunol. 2019, 10.
- Hourcade, D.; Holers, V.M.; Atkinson, J.P. The Regulators of Complement Activation (RCA) Gene Cluster. In Advances in Immunology; Dixon, F.J., Ed.; Academic Press: Cambridge, MA, USA, 1989; Volume 45, pp. 381–416.
- Liszewski, M.K.; Leung, M.; Cui, W.; Subramanian, V.B.; Parkinson, J.; Barlow, P.N.; Manchester, M.; Atkinson, J.P. Dissecting sites important for complement regulatory activity in membrane cofactor protein (MCP.; CD46). J. Biol. Chem. 2000, 275, 37692–37701.
- Krych-Goldberg, M.; Atkinson, J.P. Structure-function relationships of complement receptor type 1. Immunol. Rev. 2001, 180, 112–122.
- Jozsi, M.; Zipfel, P.F. Factor H family proteins and human diseases. Trends Immunol. 2008, 29, 380–387.
- Wu, J.; Wu, Y.-Q.; Ricklin, D.; Janssen, B.J.C.; Lambris, J.D.; Gros, P. Structure of complement fragment C3b-factor H and implications for host protection by complement regulators. Nat. Immunol. 2009, 10, 728–733.
- Ricklin, D.; Reis, E.S.; Lambris, J.D. Complement in disease: A defence system turning offensive. Nat. Rev. Nephrol. 2016, 12, 383–401.
- Coulthard, L.G.; Woodruff, T.M. Is the Complement Activation Product C3a a Proinflammatory Molecule? Re-evaluating the Evidence and the Myth. J. Immunol. 2015, 194, 3542–3548.
- Helmy, K.Y.; Katschke, K.J., Jr.; Gorgani, N.N.; Kljavin, N.M.; Elliott, J.M.; Diehl, L.; Scales, S.J.; Ghilardi, N.; van Lookeren Campagne, M. CRIg: A macrophage complement receptor required for phagocytosis of circulating pathogens. Cell 2006, 124, 915–927.
- Liu, G.; Fu, Y.; Yosri, M.; Chen, Y.; Sun, P.; Xu, J.; Zhang, M.; Sun, D.; Strickland, A.B.; Mackey, Z.B.; et al. CRIg plays an essential role in intravascular clearance of bloodborne parasites by interacting with complement. Proc. Natl. Acad. Sci. USA 2019, 116, 24214–24220.
- Vorup-Jensen, T.; Jensen, R.K. Structural Immunology of Complement Receptors 3 and 4. Front. Immunol. 2018, 9, 2716.
- Gonzalez, S.F.; Lukacs-Kornek, V.; Kuligowski, M.P.; Pitcher, L.A.; Degn, S.E.; Turley, S.J.; Carroll, M.C. Complement-Dependent Transport of Antigen into B Cell Follicles. J. Immunol. 2010, 185, 2659–2664.
- Pangburn, M.K.; Rawal, N. Structure and function of complement C5 convertase enzymes. Biochem. Soc. Trans. 2002, 30, 1006–1010.
- Rawal, N.; Pangburn, M.K. Formation of High Affinity C5 Convertase of the Classical Pathway of Complement. J. Biol. Chem. 2003, 278, 38476–38483.
- Fredslund, F.; Laursen, N.S.; Roversi, P.; Jenner, L.; Oliveira, C.L.; Pedersen, J.S.; Nunn, M.A.; Lea, S.M.; Discipio, R.; Sottrup-Jensen, L.; et al. Structure of and influence of a tick complement inhibitor on human complement component 5. Nat. Immunol. 2008, 9, 753–760.
- Bayly-Jones, C.; Bubeck, D.; Dunstone, M.A. The mystery behind membrane insertion: A review of the complement membrane attack complex. Philos Trans. R Soc. Lond. B Biol. Sci. 2017, 372, 20160221.
- Heesterbeek, D.A.C.; Angelier, M.L.; Harrison, R.A.; Rooijakkers, S.H.M. Complement and Bacterial Infections: From Molecular Mechanisms to Therapeutic Applications. J. Innate Immun. 2018, 10, 455–464.
- Klos, A.; Tenner, A.J.; Johswich, K.-O.; Ager, R.R.; Reis, E.S.; Köhl, J. The role of the anaphylatoxins in health and disease. Mol. Immunol. 2009, 46, 2753–2766.
- Hill, A.; DeZern, A.E.; Kinoshita, T.; Brodsky, R.A. Paroxysmal nocturnal haemoglobinuria. Nat. Rev. Dis. Primers 2017, 3, 17028.
- Nicholson-Weller, A.; March, J.P.; Rosenfeld, S.I.; Austen, K.F. Affected erythrocytes of patients with paroxysmal nocturnal hemoglobinuria are deficient in the complement regulatory protein, decay accelerating factor. Proc. Natl. Acad. Sci. USA 1983, 80, 5066–5070.
- Holguin, M.H.; Fredrick, L.R.; Bernshaw, N.J.; Wilcox, L.A.; Parker, C.J. Isolation and characterization of a membrane protein from normal human erythrocytes that inhibits reactive lysis of the erythrocytes of paroxysmal nocturnal hemoglobinuria. J. Clin. Invest. 1989, 84, 7–17.
- Zhang, K.; Lu, Y.; Harley, K.T.; Tran, M.-H. Atypical Hemolytic Uremic Syndrome: A Brief Review. Hematol. Rep. 2017, 9, 7053.
- Bresin, E.; Rurali, E.; Caprioli, J.; Sanchez-Corral, P.; Fremeaux-Bacchi, V.; Rodriguez de Cordoba, S.; Pinto, S.; Goodship, T.H.J.; Alberti, M.; Ribes, D.; et al. Combined complement gene mutations in atypical hemolytic uremic syndrome influence clinical phenotype. J. Am. Soc. Nephrol. 2013, 24, 475–486.
- Rodríguez de Córdoba, S.; Hidalgo, M.S.; Pinto, S.; Tortajada, A. Genetics of atypical hemolytic uremic syndrome (aHUS). Semin. Thromb. Hemost. 2014, 40, 422–430.
- Fakhouri, F.; Fremeaux-Bacchi, V.; Noel, L.H.; Cook, H.T.; Pickering, M.C. C3 glomerulopathy: A new classification. Nat. Rev. Nephrol. 2010, 6, 494–499.
- Smith, R.J.H.; Appel, G.B.; Blom, A.M.; Cook, H.T.; D’Agati, V.D.; Fakhouri, F.; Fremeaux-Bacchi, V.; Józsi, M.; Kavanagh, D.; Lambris, J.D.; et al. C3 glomerulopathy - understanding a rare complement-driven renal disease. Nat. Rev. Nephrol. 2019, 15, 129–143.
- Zhang, Y.; Meyer, N.C.; Wang, K.; Nishimura, C.; Frees, K.; Jones, M.; Katz, L.M.; Sethi, S.; Smith, R.J. Causes of alternative pathway dysregulation in dense deposit disease. Clin. J. Am. Soc. Nephrol. 2012, 7, 265–274.
- Bu, F.; Borsa, N.G.; Jones, M.B.; Takanami, E.; Nishimura, C.; Hauer, J.J.; Azaiez, H.; Black-Ziegelbein, E.A.; Meyer, N.C.; Kolbe, D.L.; et al. High-Throughput Genetic Testing for Thrombotic Microangiopathies and C3 Glomerulopathies. J. Am. Soc. Nephrol. 2016, 27, 1245–1253.
- Van Lookeren Campagne, M.; Strauss, E.C.; Yaspan, B.L. Age-related macular degeneration: Complement in action. Immunobiology 2016, 221, 733–739.
- Fritsche, L.G.; Chen, W.; Schu, M.; Yaspan, B.L.; Yu, Y.; Thorleifsson, G.; Zack, D.J.; Arakawa, S.; Cipriani, V.; Ripke, S.; et al. Seven new loci associated with age-related macular degeneration. Nat. Genet. 2013, 45, 433–439.e2.
- Ogden, C.A.; deCathelineau, A.; Hoffmann, P.R.; Bratton, D.; Ghebrehiwet, B.; Fadok, V.A.; Henson, P.M. C1q and mannose binding lectin engagement of cell surface calreticulin and CD91 initiates macropinocytosis and uptake of apoptotic cells. J. Exp. Med. 2001, 194, 781–795.
- Botto, M.; Walport, M.J. C1q, Autoimmunity and Apoptosis. Immunobiology 2002, 205, 395–406.
- Garcia, B.L.; Zwarthoff, S.A.; Rooijakkers, S.H.M.; Geisbrecht, B.V. Novel Evasion Mechanisms of the Classical Complement Pathway. J. Immunol. 2016, 197, 2051–2060.
- Jäger, U.; D’Sa, S.; Schörgenhofer, C.; Bartko, J.; Derhaschnig, U.; Sillaber, C.; Jilma-Stohlawetz, P.; Fillitz, M.; Schenk, T.; Patou, G.; et al. Inhibition of complement C1s improves severe hemolytic anemia in cold agglutinin disease: A first-in-human trial. Blood 2019, 133, 893–901.
- Ricklin, D.; Lambris, J.D. Complement in Immune and Inflammatory Disorders: Therapeutic Interventions. J. Immunol. 2013, 190, 3839.
- Wouters, D.; Zeerleder, S. Complement inhibitors to treat IgM-mediated autoimmune hemolysis. Haematologica 2015, 100, 1388–1395.
- Lintner, K.E.; Wu, Y.L.; Yang, Y.; Spencer, C.H.; Hauptmann, G.; Hebert, L.A.; Atkinson, J.P.; Yu, C.Y. Early Components of the Complement Classical Activation Pathway in Human Systemic Autoimmune Diseases. Front. Immunol. 2016, 7.
- Johnson, M.B.; Stevens, B. Pruning hypothesis comes of age. Nature 2018, 554, 438–439.
- Stevens, B.; Allen, N.J.; Vazquez, L.E.; Howell, G.R.; Christopherson, K.S.; Nouri, N.; Micheva, K.D.; Mehalow, A.K.; Huberman, A.D.; Stafford, B.; et al. The Classical Complement Cascade Mediates CNS Synapse Elimination. Cell 2007, 131, 1164–1178.
- Stephan, A.H.; Barres, B.A.; Stevens, B. The Complement System: An Unexpected Role in Synaptic Pruning During Development and Disease. Annu. Rev. Neurosci. 2012, 35, 369–389.
- Sekar, A.; Bialas, A.R.; de Rivera, H.; Davis, A.; Hammond, T.R.; Kamitaki, N.; Tooley, K.; Presumey, J.; Baum, M.; Van Doren, V.; et al. Schizophrenia risk from complex variation of complement component 4. Nature 2016, 530, 177–183.
- Schizophrenia Working Group of the Psychiatric Genomics Consortium. Biological insights from 108 schizophrenia-associated genetic loci. Nature 2014, 511, 421–427.
- Baum, M.L.; Wilton, D.K.; Muthukumar, A.; Fox, R.G.; Carey, A.; Crotty, W.; Scott-Hewitt, N.; Bien, E.; Sabatini, D.A.; Lanser, T.; et al. CUB and Sushi Multiple Domains 1 (CSMD1) opposes the complement cascade in neural tissues. bioRxiv 2020.
- Comer, A.L.; Jinadasa, T.; Sriram, B.; Phadke, R.A.; Kretsge, L.N.; Nguyen, T.P.H.; Antognetti, G.; Gilbert, J.P.; Lee, J.; Newmark, E.R.; et al. Increased expression of schizophrenia-associated gene C4 leads to hypoconnectivity of prefrontal cortex and reduced social interaction. PLoS Biol. 2020, 18, e3000604.
- Lui, H.; Zhang, J.; Makinson, S.R.; Cahill, M.K.; Kelley, K.W.; Huang, H.-Y.; Shang, Y.; Oldham, M.C.; Martens, L.H.; Gao, F.; et al. Progranulin Deficiency Promotes Circuit-Specific Synaptic Pruning by Microglia via Complement Activation. Cell 2016, 165, 921–935.
- Hong, S.; Beja-Glasser, V.F.; Nfonoyim, B.M.; Frouin, A.; Li, S.; Ramakrishnan, S.; Merry, K.M.; Shi, Q.; Rosenthal, A.; Barres, B.A.; et al. Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 2016, 352, 712–716.
- Vukojicic, A.; Delestrée, N.; Fletcher, E.V.; Pagiazitis, J.G.; Sankaranarayanan, S.; Yednock, T.A.; Barres, B.A.; Mentis, G.Z. The Classical Complement Pathway Mediates Microglia-Dependent Remodeling of Spinal Motor Circuits during Development and in SMA. Cell Rep. 2019, 29, 3087–3100.e3087.
- Carpanini, S.M.; Torvell, M.; Morgan, B.P. Therapeutic Inhibition of the Complement System in Diseases of the Central Nervous System. Front. Immunol. 2019, 10, 362.
- Vasek, M.J.; Garber, C.; Dorsey, D.; Durrant, D.M.; Bollman, B.; Soung, A.; Yu, J.; Perez-Torres, C.; Frouin, A.; Wilton, D.K.; et al. A complement–microglial axis drives synapse loss during virus-induced memory impairment. Nature 2016, 534, 538.