The increase in pandemics caused by RNA viruses of zoonotic origin highlights the urgent need for broad-spectrum antivirals against novel and re-emerging RNA viruses. Broad-spectrum anti-virals could be deployed as first-line interventions during an outbreak while virus-specific drugs and vaccines are developed and rolled out. Viruses depend on the host’s protein synthesis ma-chinery for replication. Several natural compounds that target the cellular DEAD-box RNA hel-icase eIF4A, a key component of the eukaryotic translation initiation complex eIF4F, have emerged as potential broad-spectrum antivirals. Rocaglates, a group of flavaglines of plant origin that clamp mRNAs with highly structured 5′ untranslated regions (5′UTRs) onto the surface of eIF4A through specific stacking interactions, exhibit the largest selectivity and potential therapeu-tic indices among all known eIF4A inhibitors. Their unique mechanism of action limits the inhibi-tory effect of rocaglates to the translation of eIF4A-dependent viral mRNAs and a minor fraction of host mRNAs exhibiting stable RNA secondary structures and/or polypurine sequence stretches in their 5′UTRs, resulting in minimal potential toxic side effects.
- pan-antiviral
- rocaglates
- eIF4A
- silvestrol
- CR-31-B
- Zotatifin
- translation initiation
- coronavirus
- COVID-19
1. Introduction
The frequency of infectious disease outbreaks caused by RNA viruses of zoonotic origin in human populations has been rising at an alarming rate in recent years. Prominent examples since the beginning of this millennium are severe acute respiratory syndrome coronavirus (SARS-CoV) (2002–2004), influenza A virus subtype H1N1 (2009–2010), and Middle East respiratory syndrome coronavirus (MERS-CoV) (2012-present). The causes for this intensification are many, including anthropogenic modifications of the environment, encroachment of humans into forested and other natural areas, and overall increased human contact with wildlife [1].
The ongoing SARS-CoV-2 pandemic is the latest example of the widespread and damaging effects such outbreaks can have on a global scale. In just the past three decades, such events have resulted in millions of excess deaths and in economic losses totaling billions of USD [2][3][4]. Preventing new outbreaks from occurring through continuous and coordinated surveillance remains a top public health priority, but developing strategies to mitigate the effects of an outbreak once it is detected, and while a vaccine is developed, has taken on an added sense of urgency [5].
The ongoing SARS-CoV-2 pandemic is the latest example of the widespread and damaging effects such outbreaks can have on a global scale. In just the past three decades, such events have resulted in millions of excess deaths and in economic losses totaling billions of USD [2–4]. Preventing new outbreaks from occurring through continuous and coordinated surveillance remains a top public health priority, but developing strategies to mitigate the effects of an outbreak once it is detected, and while a vaccine is developed, has taken on an added sense of urgency [5].
In parallel to the increase in the incidence of infectious diseases, there has been a gradual shift toward more outbreaks being of viral origin rather than caused by other infectious organisms such as bacteria, protozoa, parasites, or fungi [6]. Historically, viruses constituted about one-tenth of all novel human pathogens of zoonotic origin but since 1980 that proportion has increased to about two-thirds [7]. Viruses replicate with extremely high mutation rates compared to other microorganisms, allowing them to rapidly adapt to new hosts and evolve resistance to vaccines and antiviral drugs [8]. Moreover, among viruses, RNA viruses exhibit the highest mutation rates compared to DNA viruses, which gives them an evolutionary advantage that results in RNA viruses accounting for over 80% of the zoonotic viral burden and a disproportionate contribution to the total burden of zoonotic human pathogenesis [9]. Indeed, all major epidemic and pandemic outbreaks since 2000 have been caused by RNA viruses (Table 1). This dominance is most likely not just a result of their high genetic plasticity but also of their high human-to-human transmissibility, especially in cases where RNA viruses can be transmitted by aerosols [10].
Table 1.
Major epidemics and pandemics of zoonotic origin since 2000 (Source: WHO). Abbreviations: COVID-19—coronavirus disease 2019; SARS-CoV-2—severe acute respiratory syndrome coronavirus 2; ZIKV—Zika virus; CHIKV—Chikungunya virus; EBOV—Ebola virus; IAV/H1N1—influenza A virus subtype H1N1.
|
Years |
Disease |
Causative Agent |
Epidemic/Pandemic |
Taxonomic Family |
|
2019–present |
COVID-19 |
SARS-CoV-2 |
Pandemic |
Coronaviridae |
|
2015–present |
Zika virus disease |
ZIKV |
Epidemic |
Flaviviridae |
|
2015/2016 |
Chikungunya fever |
CHIKV |
Epidemic |
Togaviridae |
|
2014–2016 |
Ebola hemorrhagic fever |
EBOV |
Epidemic |
Filoviridae |
|
2012–present |
Middle East respiratory syndrome |
MERS-CoV |
Epidemic |
Coronaviridae |
|
2009/2010 |
Influenza |
IAV/H1N1 |
Pandemic |
Orthomyxoviridae |
|
2002/2003 |
Severe acute respiratory syndrome |
SARS-CoV |
Epidemic |
Coronaviridae |
Given the disproportionate contribution of RNA viruses to human infectious disease of zoonotic origin and their dominant role in causing major epidemics and pandemics over the past 20 years, developing antiviral therapeutics targeting RNA viruses should be a priority in the quest to curb the spread of future emerging and re-emerging RNA virus outbreaks. Accurate predictions of which RNA viruses will cause future outbreaks is however impossible due to the multiplicity of potential animal hosts and the genetically highly heterogeneous makeup of the virus strains present in these hosts [11]. Ideally therefore, antivirals developed for use against future outbreaks of zoonotic RNA viruses would have to be effective against positive (+)- and negative (−)-stranded RNA viruses across as many virus families as possible.
Antivirals can target either the virus or the host. Virus-specific or direct-targeting antiviral strategies, which include neutralizing antibodies targeting surface antigens of the virus itself, compounds targeting virus–receptor interactions, fusion/budding inhibitors, and viral protease inhibitors [12], are by definition directed against known viral strains, and their development can only be promoted to a preliminary stage before an outbreak happens. Nonspecific virus-targeting antivirals, which consist mostly of nucleoside analogs that inhibit the viral RNA polymerase machinery [12], have found broader application because they can be developed a priori, but their efficacies and safety profiles have been inconsistent and not directly transferable among viral strains. The biggest drawback of direct-targeting antivirals however has been the high selective pressure they exert on the RNA viruses themselves. This results in enhanced mutation rates and translates into unpredictable “moving goalposts” for drug development and, in more extreme cases, gives rise to escape mutants that can render antivirals altogether ineffective over time [13][14][15].
Antivirals can target either the virus or the host. Virus-specific or direct-targeting antiviral strategies, which include neutralizing antibodies targeting surface antigens of the virus itself, compounds targeting virus–receptor interactions, fusion/budding inhibitors, and viral protease inhibitors [12], are by definition directed against known viral strains, and their development can only be promoted to a preliminary stage before an outbreak happens. Nonspecific virus-targeting antivirals, which consist mostly of nucleoside analogs that inhibit the viral RNA polymerase machinery [12], have found broader application because they can be developed a priori, but their efficacies and safety profiles have been inconsistent and not directly transferable among viral strains. The biggest drawback of direct-targeting antivirals however has been the high selective pressure they exert on the RNA viruses themselves. This results in enhanced mutation rates and translates into unpredictable “moving goalposts” for drug development and, in more extreme cases, gives rise to escape mutants that can render antivirals altogether ineffective over time [13–15].
By contrast, host-targeting antiviral strategies, including host receptor inhibitors, endosomal pathway inhibitors, host protease inhibitors, modulators of lipid metabolism, modulators of innate immune response or assorted nuclear signaling pathway modulators [16], minimize the risk of viral mutations by eliminating any direct selective pressure on the viruses. A potential drawback of host-targeting antivirals, however, resides in the risk of substantial side effects caused by interfering with the normal function of the targeted host factors and pathways. Consequently, host-targeted antivirals tend to have narrow therapeutic windows and are complex to manage from a clinical perspective [15].
By contrast, host-targeting antiviral strategies, including host receptor inhibitors, endosomal pathway inhibitors, host protease inhibitors, modulators of lipid metabolism, modulators of innate immune response or assorted nuclear signaling pathway modulators [16], minimize the risk of viral mutations by eliminating any direct selective pressure on the viruses. A potential drawback of host-targeting antivirals, however, resides in the risk of substantial side effects caused by interfering with the normal function of the targeted host factors and pathways [11]. Consequently, host-targeted antivirals tend to have narrow therapeutic windows and are complex to manage from a clinical perspective [15].
One host mechanism essential to viral proliferation is translation. RNA viruses do not encode their own translational machinery, rendering them dependent on host protein synthesis. The eukaryotic translational machinery has long been targeted in the context of cancer because aberrant mRNA translation and high expression levels of oncogenes are two hallmarks of many neoplasias [17]. Targeting protein synthesis to inhibit viral proliferation has only been proposed more recently as an attractive therapeutic option to treat viral or bacterial infections [18][19]. The ongoing SARS-CoV-2 pandemic has further accelerated the clinical development of such antivirals through repurposing compounds already in development to inhibit host translation factors in the context of cancer [20][21][22][23]. Importantly, the potential broad-spectrum application of host-specific translational inhibitors is a crucial argument for their development. Such pan-antivirals could be deployed as first-line drugs in the event of an epidemic or pandemic outbreak caused by a novel virus for which there are no direct-targeting antivirals available.
One host mechanism essential to viral proliferation is translation. RNA viruses do not encode their own translational machinery, rendering them dependent on host protein synthesis. The eukaryotic translational machinery has long been targeted in the context of cancer because aberrant mRNA translation and high expression levels of oncogenes are two hallmarks of many neoplasias [17]. Targeting protein synthesis to inhibit viral proliferation has only been proposed more recently as an attractive therapeutic option to treat viral or bacterial infections [18,19]. The ongoing SARS-CoV-2 pandemic has further accelerated the clinical development of such antivirals through repurposing compounds already in development to inhibit host translation factors in the context of cancer [20–23]. Importantly, the potential broad-spectrum application of host-specific translational inhibitors is a crucial argument for their development. Such pan-antivirals could be deployed as first-line drugs in the event of an epidemic or pandemic outbreak caused by a novel virus for which there are no direct-targeting antivirals available.
Recent advances in the development of compounds to target the cellular, cap-dependent DEAD-box RNA helicase eIF4A, an essential factor in viral protein synthesis, have underscored the potential of targeting this translational host factor as an antiviral strategy [24]. A large number of RNA viruses depend on eIF4A to translate their mRNAs because the complex structure of their 5′ untranslated regions (5′UTRs) requires the helicase activity of eIF4A to form the 43S-preinitiation complex (43S-PIC) during translation initiation [25]. In vitro, ex vivo, and in vivo inhibition of eIF4A with small natural compounds has been shown to prevent replication of RNA viruses, including corona-, picorna-, flavi-, filo-, hepe-, toga-, arena-, nairo-, and bunyaviruses [26][27].
Recent advances in the development of compounds to target the cellular, cap-dependent DEAD-box RNA helicase eIF4A, an essential factor in viral protein synthesis, have underscored the potential of targeting this translational host factor as an antiviral strategy [24]. A large number of RNA viruses depend on eIF4A to translate their mRNAs because the complex structure of their 5′ untranslated regions (5′UTRs) requires the helicase activity of eIF4A to form the 43S-preinitiation complex (43S-PIC) during translation initiation [25]. In vitro, ex vivo, and in vivo inhibition of eIF4A with small natural compounds has been shown to prevent replication of RNA viruses, including corona-, picorna-, flavi-, filo-, hepe-, toga-, arena-, nairo-, and bunyaviruses [26,27].
A very promising class of eIF4A inhibitors are rocaglates, a group of flavaglines that target eIF4A with high specificity [28][29]. The resulting high potencies and optimal selectivity indices compared to all other known eIF4A inhibitors make rocaglates ideal candidates for further preclinical and clinical development as pan-antivirals.
A very promising class of eIF4A inhibitors are rocaglates, a group of flavaglines that target eIF4A with high specificity [28,29]. The resulting high potencies and optimal selectivity indices compared to all other known eIF4A inhibitors make rocaglates ideal candidates for further preclinical and clinical development as pan-antivirals.
2. The Role of the DEAD-Box RNA Helicase eIF4A in RNA Virus Translation
All known viruses usurp the translational machinery of their host cells to synthesize large amounts of viral proteins in a short period of time. To achieve this, viral mRNAs compete with the host’s own mRNAs for access to the necessary factors of the eukaryotic translation initiation apparatus, a complex machinery comprising at least twelve different components [30]. Indeed, all viruses, but RNA viruses in particular, have developed several sophisticated strategies to direct the host’s translation machinery to preferentially synthesize viral proteins over the host’s own proteins [31][32][33]. The two translation mechanisms mainly used by RNA viruses are cap-dependent translation and internal ribosome entry site (IRES)-dependent translation.
All known viruses usurp the translational machinery of their host cells to synthesize large amounts of viral proteins in a short period of time. To achieve this, viral mRNAs compete with the host’s own mRNAs for access to the necessary factors of the eukaryotic translation initiation apparatus, a complex machinery comprising at least twelve different components [30]. Indeed, all viruses, but RNA viruses in particular, have developed several sophisticated strategies to direct the host’s translation machinery to preferentially synthesize viral proteins over the host’s own proteins [31–33]. The two translation mechanisms mainly used by RNA viruses are cap-dependent translation and internal ribosome entry site (IRES)-dependent translation.
Cap-dependent translation follows a precise sequence of events orchestrated by different eukaryotic initiation factors (eIFs) [34]. First, the mRNA is recruited to the heterotrimeric eIF4F complex [35]. This complex consists of the cap-binding protein eIF4E, eIF4A, and the scaffold protein eIF4G that binds to the poly (A)-binding protein (PABP) and eIF3, another initiation factor [36][37]. Binding of eIF4F to the cap structure leads to cyclization of the mRNA [38]. Next, and in order for the 40S ribosomal subunit to gain access to the highly structured viral 5′UTRs, eIF4A springs into action [39]. The helicase unwinds RNA secondary structures in the 5´UTR and removes adherent proteins to generate an unstructured region that allows stable binding of the 43S-PIC [40][41]. This complex scans the 5´UTR to identify the AUG start codon where formation of the elongation-competent 80S complex takes place followed by protein synthesis [41][42][43][44] (Figure 1).
Cap-dependent translation follows a precise sequence of events orchestrated by different eukaryotic initiation factors (eIFs) [34]. First, the mRNA is recruited to the heterotrimeric eIF4F complex [35]. This complex consists of the cap-binding protein eIF4E, eIF4A, and the scaffold protein eIF4G that binds to the poly (A)-binding protein (PABP) and eIF3, another initiation factor [36,37]. Binding of eIF4F to the cap structure leads to cyclization of the mRNA [38]. Next, and in order for the 40S ribosomal subunit to gain access to the highly structured viral 5′UTRs, eIF4A springs into action [39]. The helicase unwinds RNA secondary structures in the 5´UTR and removes adherent proteins to generate an unstructured region that allows stable binding of the 43S-PIC [40,41]. This complex scans the 5´UTR to identify the AUG start codon where formation of the elongation-competent 80S complex takes place followed by protein synthesis [38,41–44] (Figure 1).
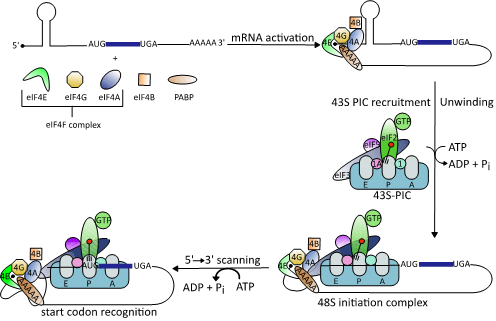
Figure 1.
Schematic illustration of the eukaryotic translation initiation mechanism (modified from [38]). Binding of the heterotrimeric eIF4F complex to the 5′ cap structure of mRNAs is followed by unwinding of stable RNA secondary structures by the DEAD-box RNA helicase eIF4A. This enables binding of the 43S preinitiation complex (PIC), which scans down the 5´-untranslated regions (5’UTRs) to identify the start codon AUG.
In IRES-dependent translation, the 40S ribosomal subunit is recruited directly to the mRNA start codon by binding to the secondary structure of an IRES [45]. Most viral IRES-dependent translation mechanisms have been categorized into one of four classes based on the RNA structures involved and the initiation factors they recruit [46]. In classes I to III, the IRES is located in the 5′UTR and translation initiation depends on eIFs, while class IV IRES are located in intergenic regions and do not recruit any eIFs. Class I and II IRES contain simple short and long hairpin structures while class III IRES contain more complex, knotted secondary structures. Class I and II IRES-dependent translation requires a combination of eIF4G, eIF3, eIF2, and eIF4A, while class III does not use eIF4A to assemble the 40S initiation complex [46][47][48]. Finally, ribosome recruitment occurs upstream of the start codon in class I IRES-dependent translation and requires 5′ to 3′ scanning to reach the start codon, while the translation complex binds directly to the start codon in class II IRES-dependent translation.
In IRES-dependent translation, the 40S ribosomal subunit is recruited directly to the mRNA start codon by binding to the secondary structure of an IRES [45]. Most viral IRES-dependent translation mechanisms have been categorized into one of four classes based on the RNA structures involved and the initiation factors they recruit [46]. In classes I to III, the IRES is located in the 5′UTR and translation initiation depends on eIFs, while class IV IRES are located in intergenic regions and do not recruit any eIFs. Class I and II IRES contain simple short and long hairpin structures while class III IRES contain more complex, knotted secondary structures. Class I and II IRES-dependent translation requires a combination of eIF4G, eIF3, eIF2, and eIF4A, while class III does not use eIF4A to assemble the 40S initiation complex [46–48]. Finally, ribosome recruitment occurs upstream of the start codon in class I IRES-dependent translation and requires 5′ to 3′ scanning to reach the start codon, while the translation complex binds directly to the start codon in class II IRES-dependent translation [46].
The translation initiation factor eIF4A is a DEAD-box RNA helicase, a group of ATP-dependent eukaryotic RNA helicases named after the conserved amino acid sequence Asp-Glu-Ala-Asp (D-E-A-D) [49]. There are three paralogs of eIF4A in mammals: eIF4A1 (DDX2A), eIF4A2 (DDX2B), and eIF4A3 (DDX48). While eIF4A1 and eIF4A2 have a sequence identity of 90–95%, eIF4A3 shares only ~60% sequence identity with eIF4A1 [50]. Functionally, eIF4A3 differs from eIF4A1 and eIF4A2. eIF4A3 is involved in the assembly of the Exon Junction Complex, which coordinates splicing of pre-mRNAs [50]. By contrast, eIF4A1 and eIF4A2 exhibit equivalent biochemical activities but differ significantly in biological function and expression levels in vivo. eIF4A1 is present in almost all tissues during active cell growth, whereas eIF4A2 is mainly produced in organs with low proliferation rates [51]. In addition, when eIF4A1 is suppressed, eIF4A2 levels rise, but the loss of eIF4A1 is not fully compensated. Consequently, eIF4A1 but not eIF4A2 is essential for cell survival.
The translation initiation factor eIF4A is a DEAD-box RNA helicase, a group of ATP-dependent eukaryotic RNA helicases named after the conserved amino acid sequence Asp-Glu-Ala-Asp (D-E-A-D) [49]. There are three paralogs of eIF4A in mammals: eIF4A1 (DDX2A), eIF4A2 (DDX2B), and eIF4A3 (DDX48). While eIF4A1 and eIF4A2 have a sequence identity of 90–95%, eIF4A3 shares only ~60% sequence identity with eIF4A1 [50]. Functionally, eIF4A3 differs from eIF4A1 and eIF4A2. eIF4A3 is involved in the assembly of the Exon Junction Complex, which coordinates splicing of pre-mRNAs [50]. By contrast, eIF4A1 and eIF4A2 exhibit equivalent biochemical activities but differ significantly in biological function and expression levels in vivo. eIF4A1 is present in almost all tissues during active cell growth, whereas eIF4A2 is mainly produced in organs with low proliferation rates [51]. In addition, when eIF4A1 is suppressed, eIF4A2 levels rise, but the loss of eIF4A1 is not fully compensated. Consequently, eIF4A1 but not eIF4A2 is essential for cell survival.
All DEAD-box RNA helicases contain two RecA-like domains that are connected by a flexible linker [52]. In the absence of an RNA substrate or ATP, the two domains adopt an inactive open conformation [53][54]. Binding to RNA and ATP leads to closure of the gap between the two domains (Figure 2). In this closed, active conformation, the conserved motifs of the two domains form an interface exhibiting ATPase and helicase activities. Following ATP hydrolysis, the gap between the two domains re-opens to allow the release of the unwound RNA.
All DEAD-box RNA helicases contain two RecA-like domains that are connected by a flexible linker [52]. In the absence of an RNA substrate or ATP, the two domains adopt an inactive open conformation [53,54]. Binding to RNA and ATP leads to closure of the gap between the two domains (Figure 2). In this closed, active conformation, the conserved motifs of the two domains form an interface exhibiting ATPase and helicase activities. Following ATP hydrolysis, the gap between the two domains re-opens to allow the release of the unwound RNA.
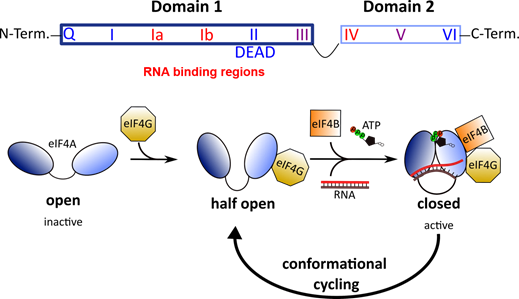
Figure 2. Top: Architecture of DEAD-box RNA helicases. Two RecA-like domains are connected by a flexible linker to form the helicase core. This core consists of 9 conserved motifs that are involved in ATP binding and hydrolysis (blue) and RNA binding (red). Bottom: Conformational cycling of eIF4A. Binding of eIF4G enables eIF4A to switch from the open to the half-open state. In the presence of eIF4B, ATP and an RNA substrate, eIF4A can undergo conformational cycling and alternate between the active-closed and half-open state to enable helicase and ATPase activity.
Top: Architecture of DEAD-box RNA helicases. Two RecA-like domains are connected by a flexible linker to form the helicase core. This core consists of 9 conserved motifs that are involved in ATP binding and hydrolysis (blue) and RNA binding (red) [50]. Bottom: Conformational cycling of eIF4A. Binding of eIF4G enables eIF4A to switch from the open to the half-open state. In the presence of eIF4B, ATP and an RNA substrate, eIF4A can undergo conformational cycling and alternate between the active-closed and half-open state to enable helicase and ATPase activity [53].
While eIF4A is the main helicase responsible for unwinding RNA secondary structures in 5′UTRs, several other RNA helicases play essential roles during translation [55]. Among them, the RNA helicase DEAD-box polypeptide 3 (DDX3) was reported to facilitate translation of complex secondary RNA structures in general as well as of secondary structures specifically associated with the 7-methylguanylate structure (m
7GTP) of the RNA cap [46]. DDX3 also plays a role equivalent to that of eIF4A in class I and II IRES-dependent translation [46], and its essential role in viral RNA translation has been documented for a number of viruses, including the RNA viruses Japanese encephalitis virus, Dengue virus (DENV), and West Nile virus [56][57][58][59]. Several studies have further shown the broad-spectrum antiviral potential of targeting DDX3 [60][61][62].
GTP) of the RNA cap [46]. DDX3 also plays a role equivalent to that of eIF4A in class I and II IRES-dependent translation [46], and its essential role in viral RNA translation has been documented for a number of viruses, including the RNA viruses Japanese encephalitis virus, Dengue virus (DENV), and West Nile virus [56–59]. Several studies have further shown the broad-spectrum antiviral potential of targeting DDX3 [60–62].
3. eIF4A Inhibitors
A number of natural products that inhibit protein synthesis in eukaryotic cells, and eIF4A in particular, have been described, and their number continues to grow [63][64] (Figure 3). Originally, eIF4A inhibitors were identified as potential antitumor therapeutics [65][66]. In addition to its ATP-binding pocket, eIF4A has a nucleic acid-binding region where RNA substrates bind via their phosphate backbone in a sequence-independent manner, providing several possible interaction surfaces for inhibitors to bind to the eIF4A–RNA complex [67]. High-throughput screens for eukaryotic translation inhibitors resulted in the identification of three natural substances—hippuristanol, pateamine A (PatA), and silvestrol—that differ substantially in their chemical structures [68][69][70].
A number of natural products that inhibit protein synthesis in eukaryotic cells, and eIF4A in particular, have been described, and their number continues to grow [63,64] (Figure 3). Originally, eIF4A inhibitors were identified as potential antitumor therapeutics [65,66]. In addition to its ATP-binding pocket, eIF4A has a nucleic acid-binding region where RNA substrates bind via their phosphate backbone in a sequence-independent manner, providing several possible interaction surfaces for inhibitors to bind to the eIF4A–RNA complex [67]. High-throughput screens for eukaryotic translation inhibitors resulted in the identification of three natural substances—hippuristanol, pateamine A (PatA), and silvestrol—that differ substantially in their chemical structures [68–70].
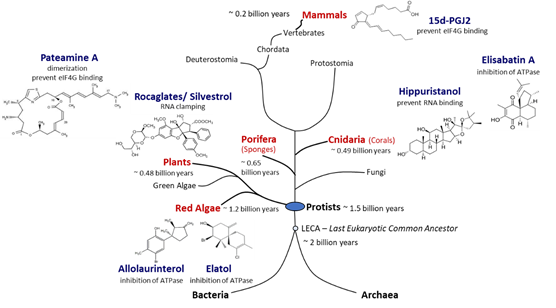
Figure 3.
Natural producers of molecules with eIF4A inhibitory activity. Compounds with eIF4A inhibitory activity can be isolated from red algae, plants, sponges, corals, and mammals. Red algae are the phylogenetically most ancient phylum of multicellular eukaryotes for which putative eIF4A inhibitors could be identified. Porifera are the most primordial multicellular animals, and cnidaria are the first animals that developed tissues. Plants and mammals are terrestrial producers of eIF4A inhibitors. Potent eIF4A inhibition has been demonstrated for rocaglates, pateamine A, and hippuristanol. The chemical structures of the inhibitors are diverse and some compounds appear to be straightforward derivatives of other bio-molecules such as sterols in case of hippuristanol or prostaglandins in case of 15d-PGJ2. Although eIF4A inhibitors have not yet been identified in fungi, blocking of eIF4A appears to be a widespread mechanism in eukaryotes.
Hippuristanol is a polyhydroxysteroid found in the golden fan coral
Isis hippuris
(Figure 3). Hippuristanol interacts with the C-terminal domain of eIF4A via motifs V and VI (Figure 2), preventing the binding of RNA [71]. Through this allosteric inhibition, eIF4A is fixed in the closed conformation and cannot release the unwound RNA substrate [72]. Hippuristanol is a selective inhibitor of eIF4A due to the high sequence variation of motifs V and VI across DEAD-box helicases [72]. In contrast to RNA binding, the binding of ATP can take place in the presence of the inhibitor because the N-terminal domain of eIF4A is not affected by hippuristanol [71].
The antiviral activity of hippuristanol has been documented against several viruses such as the encephalomyocarditis virus (EMCV) and the norovirus, two positive-stranded RNA viruses, and human T cell leukemia virus type 1 (HTLV-1), a retrovirus [73][74].
The antiviral activity of hippuristanol has been documented against several viruses such as the encephalomyocarditis virus (EMCV) and the norovirus, two positive-stranded RNA viruses, and human T cell leukemia virus type 1 (HTLV-1), a retrovirus [70,73,74].
PatA is a macrolide isolated from the encrusting marine sponge
Mycale hentscheli (Figure 3) that induces dimerization of eIF4A and RNA [75][76]. PatA disrupts interaction with eIF4G and reduces levels of eIF4A present in the eIF4F complex [75], which in turn may affect the assembly of the 43S-PIC. In contrast to hippuristanol, PatA only binds free eIF4A, suggesting that the binding site for PatA likely occurs at the interface of the eIF4A N- and C-terminals domains, two domains rendered inaccessible once the eIF4F complex has formed. PatA has been shown to have antiviral activity against influenza A virus, a negative-stranded RNA virus [77].
(Figure 3) that induces dimerization of eIF4A and RNA [69,75,76]. PatA disrupts interaction with eIF4G and reduces levels of eIF4A present in the eIF4F complex [68,75], which in turn may affect the assembly of the 43S-PIC. In contrast to hippuristanol, PatA only binds free eIF4A, suggesting that the binding site for PatA likely occurs at the interface of the eIF4A N- and C-terminals domains, two domains rendered inaccessible once the eIF4F complex has formed [66]. PatA has been shown to have antiviral activity against influenza A virus, a negative-stranded RNA virus [77].
Silvestrol is a rocaglate isolated from Asian mahogany plants of the genus
Aglaia (Figure 3). A characteristic feature of silvestrol and its diastereomer episilvestrol is the presence of a 1,4-dioxane moiety linked to their A rings (Figures 3 and 4). Since the first rocaglate, rocaglamide A (RocA), was isolated and its chemical structure solved, over 200 rocaglates have been identified [78] and isolated, including silvestrol and episilvestrol in 2004 [79][80]. A chemical synthesis route of RocA was published in 1990 that allows the control of the absolute stereochemistry of the molecule class [81]. Since then, the synthesis has been further optimized and expanded to include a large number of modified rocaglates [82]. An example of such a non-naturally occurring rocaglate is CR-31-B. The synthesis of CR-31-B results in a racemic mixture of two enantiomers, of which only the (−)-enantiomer is biologically active [83][84].
[28] (Figure 3). A characteristic feature of silvestrol and its diastereomer episilvestrol is the presence of a 1,4-dioxane moiety linked to their A rings (Figures 3 and 4) [29]. Since the first rocaglate, rocaglamide A (RocA), was isolated and its chemical structure solved, over 200 rocaglates have been identified [78] and isolated, including silvestrol and episilvestrol in 2004 [79,80]. A chemical synthesis route of RocA was published in 1990 that allows the control of the absolute stereochemistry of the molecule class [81]. Since then, the synthesis has been further optimized and expanded to include a large number of modified rocaglates [82]. An example of such a non-naturally occurring rocaglate is CR-31-B. The synthesis of CR-31-B results in a racemic mixture of two enantiomers, of which only the (−)-enantiomer is biologically active [83,84].
The antiviral activity of natural and synthetic rocaglates has been well documented (see Section 5 below).
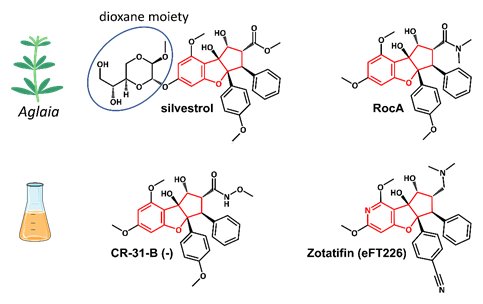
Figure 4.
Structures of the natural rocaglates silvestrol and rocaglamide A (RocA) and the synthetic rocaglates CR-31-B (-) and Zotatifin (eFT226). The typical cyclopenta[
b
]benzofurane skeleton is indicated in red. The blue ring shows the dioxane moiety that is only found in silvestrol and its diastereomer episilvestrol.
In addition to the three best-known classes of eIF4A inhibitors, other low-molecular-weight compounds have been shown to inhibit the helicase, although the specificity and selectivity of several of them remains to be established, and their potential antiviral activity has not yet been determined [85]. The list includes allolaurinterol, elatol, elisabatin A, 6-aminocholestanol, sanguinarine, and the prostaglandin 15d-PGJ2 [86][87][88][89][90]. Allolaurinterol and elatol are found in red algae and silvestrol and other rocaglates in plants, all within the supergroup
In addition to the three best-known classes of eIF4A inhibitors, other low-molecular-weight compounds have been shown to inhibit the helicase, although the specificity and selectivity of several of them remains to be established, and their potential antiviral activity has not yet been determined [85]. The list includes allolaurinterol, elatol, elisabatin A, 6-aminocholestanol, sanguinarine, and the prostaglandin 15d-PGJ2 [86–90]. Allolaurinterol and elatol are found in red algae and silvestrol and other rocaglates in plants, all within the supergroup
Archaeplastida
. By contrast, hippuristanol and elisabatin A have been isolated from cnidaria (corals) and pateamine A from porifera (sponges) species, both in the supergroup
Opisthokonta. Finally, 15d-PGJ2 is an example of an eIF4A inhibitor that is produced in mammalian (human) cells [88]. Considering that eIF4A, the prototype of DEAD-box RNA helicases, is an evolutionarily ancient and highly conserved enzyme, the widespread occurrence of chemically diverse eIF4A inhibitors across eukaryal supergroups suggests that the potential to interfere with eIF4A activity evolved independently as an advantageous antagonistic principle in eukaryal evolution (Figure 3).
. Finally, 15d-PGJ2 is an example of an eIF4A inhibitor that is produced in mammalian (human) cells [88]. Considering that eIF4A, the prototype of DEAD-box RNA helicases [49], is an evolutionarily ancient and highly conserved enzyme, the widespread occurrence of chemically diverse eIF4A inhibitors across eukaryal supergroups suggests that the potential to interfere with eIF4A activity evolved independently as an advantageous antagonistic principle in eukaryal evolution (Figure 3).
4. Mechanism of Action of Rocaglates
Rocaglates are a group of flavaglines that contain a characteristic cyclopenta[
b]benzofuran structure (Figure 4).
]benzofuran structure (Figure 4) [28,29].
Initial binding studies of eIF4A with several rocaglates—and the more recent elucidation of the crystal structure of a truncated version of human eIF4A (PDB: 5ZC9) in complex with RocA, polypurine RNA, and AMP-PNP, a non-hydrolyzable ATP analogue—have revealed that rocaglates reversibly clamp the RNA–helicase complex (Figure 5) [91][92]. Specifically, RocA forms stable π–π interactions with the phenylalanine residue at position 163 (Phe163) and two consecutive purine bases (5′-AG) in the eIF4A-(AG)
Initial binding studies of eIF4A with several rocaglates—and the more recent elucidation of the crystal structure of a truncated version of human eIF4A (PDB: 5ZC9) in complex with RocA, polypurine RNA, and AMP-PNP, a non-hydrolyzable ATP analogue—have revealed that rocaglates reversibly clamp the RNA–helicase complex (Figure 5) [65,91,92]. Specifically, RocA forms stable π–π interactions with the phenylalanine residue at position 163 (Phe163) and two consecutive purine bases (5′-AG) in the eIF4A-(AG)
5RNA complex. Additional hydrogen bonds with Gln195 and Asp198 of eIF4A, as well as with the N7 nitrogen of the guanine base of the RNA substrate, stabilize the RocA–eIF4A–RNA complex [91]. The co-crystal structure has provided the foundation for the structure-based development of new potential eIF4A inhibitors. Comparative in silico docking analysis of silvestrol and CR-31-B (-), two rocaglates with similar antiviral activities, indicates subtle differences in the binding mode between dioxane-containing silvestrol and rocaglates lacking the dioxane moiety. In silico docking results suggest an expanded interface involving contacts of the dioxane moiety of silvestrol to nearby arginine residues of eIF4A (Figure 5). This differential interaction with the eIF4A–RNA complex might explain why “larger” dioxane-containing rocaglates can clamp eIF4A–RNA complexes containing RNA substrates with short hairpin structures, while rocaglates without a dioxane moiety strictly require RNA substrates with unstructured polypurine sequences for complex formation. Ligand-based optimization studies should help further to optimize the clamping characteristics of novel rocaglates [93].
RNA complex. Additional hydrogen bonds with Gln195 and Asp198 of eIF4A, as well as with the N7 nitrogen of the guanine base of the RNA substrate, stabilize the RocA–eIF4A–RNA complex [91]. The co-crystal structure has provided the foundation for the structure-based development of new potential eIF4A inhibitors. Comparative in silico docking analysis of silvestrol and CR-31-B (-), two rocaglates with similar antiviral activities, indicates subtle differences in the binding mode between dioxane-containing silvestrol and rocaglates lacking the dioxane moiety [83]. In silico docking results suggest an expanded interface involving contacts of the dioxane moiety of silvestrol to nearby arginine residues of eIF4A (Figure 5). This differential interaction with the eIF4A–RNA complex might explain why “larger” dioxane-containing rocaglates can clamp eIF4A–RNA complexes containing RNA substrates with short hairpin structures, while rocaglates without a dioxane moiety strictly require RNA substrates with unstructured polypurine sequences for complex formation [83]. Ligand-based optimization studies should help further to optimize the clamping characteristics of novel rocaglates [23,93].
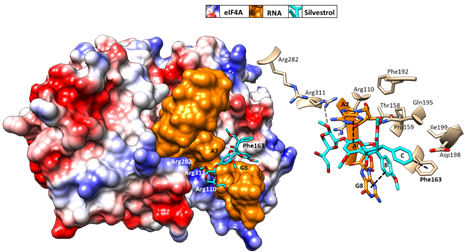
Figure 5. Predicted binding mode of silvestrol using structure-based computational modeling of silvestrol onto the eIF4A–RNA complex (PDB: 5ZC9). The dioxane moiety of silvestrol has the potential to form additional contacts with arginine residues on the surface of eIF4A and may thus bridge over the RNA substrate to tightly clamp the RNA onto eIF4A. UCSF (University of California, San Francisco) Chimera was used for graphical illustration and electrostatic surface coloring of eIF4A (blue: positive charged, red: negative charged).
Predicted binding mode of silvestrol using structure-based computational modeling of silvestrol onto the eIF4A–RNA complex (PDB: 5ZC9). The dioxane moiety of silvestrol has the potential to form additional contacts with arginine residues on the surface of eIF4A and may thus bridge over the RNA substrate to tightly clamp the RNA onto eIF4A [83]. UCSF (University of California, San Francisco) Chimera was used for graphical illustration and electrostatic surface coloring of eIF4A (blue: positive charged, red: negative charged).
DDX3, the second DEAD-box RNA helicase targeted by rocaglates [94], interacts with multiple viral components and affects several processes, including translation [95]. As a result, dual targeting of DDX3 and eIF4A might enhance the broad-spectrum antiviral effect of rocaglates. Mechanistically, there is no evidence for a π–π-stacking interaction with an aromatic residue of DDX3 that may resemble the ring C stack with Phe163 of eIF4A. However, Gln360 of DDX3, analogous to Gln195 of eIF4A, forms an essential hydrogen bond with the rocaglate [94].
DDX3, the second DEAD-box RNA helicase targeted by rocaglates [94], interacts with multiple viral components and affects several processes, including translation [95]. As a result, dual targeting of DDX3 and eIF4A might enhance the broad-spectrum antiviral effect of rocaglates. Mechanistically, there is no evidence for a π–π-stacking interaction with an aromatic residue of DDX3 that may resemble the ring C stack with Phe163 of eIF4A. However, Gln360 of DDX3, analogous to Gln195 of eIF4A, forms an essential hydrogen bond with the rocaglate [94].
At the cellular level, ribosome profiling experiments have revealed the effects of eIF4A inhibition by rocaglates on the cell’s transcriptional program. Several studies showed that the global cellular effects of eIF4A inhibition are limited to about 300 cellular mRNAs [96][97][. Among the affected mRNAs, proto-oncogenic mRNAs with relatively long and structured 5′UTRs prevailed, which explains the strong antitumor effects of rocaglates. This mRNA selectivity also might explain the low toxicity of rocaglates in primary cells and animals [98]. The cytotoxic effects of rocaglates observed in primary cells might be correlated with cellular proliferative activity [99].
At the cellular level, ribosome profiling experiments have revealed the effects of eIF4A inhibition by rocaglates on the cell’s transcriptional program. Several studies showed that the global cellular effects of eIF4A inhibition are limited to about 300 cellular mRNAs [96,97]. Among the affected mRNAs, proto-oncogenic mRNAs with relatively long and structured 5′UTRs prevailed, which explains the strong antitumor effects of rocaglates [28,79]. This mRNA selectivity also might explain the low toxicity of rocaglates in primary cells and animals [98]. The cytotoxic effects of rocaglates observed in primary cells might be correlated with cellular proliferative activity [99].