Glycogen synthase kinase-3 (GSK-3) is a central player in regulating mood behavior, cognitive functions, and neuron viability. Indeed, many targets controlled by GSK-3 are critically involved in progressing neuron deterioration and disease pathogenesis.
- GSK-3
- neurodegeneration
- microtubules
- mTOR
- autophagy
- lysosome
- mitochondria
- GSK-3 inhibitors
1. GSK-3—A Story of Two Isozymes
Glycogen synthase kinase-3 (GSK-3) is a highly conserved protein serine/threonine kinase that plays a central role in a wide variety of cellular processes concerned with coordinating catabolic and anabolic pathways and regulating cellular fate and cell growth. GSK-3 targeted phosphorylation typically inhibits the activity of the substrate, leading to attenuation of the signaling pathway. GSK-3 thus functions as a suppressor of hormone/growth factor-induced signaling cascades. For example, GSK-3 inhibits insulin signaling through the phosphorylation of glycogen synthase and the insulin receptor substrates, IRS-1/IRS-2, where the former leads to inhibition of glycogen synthesis, and the latter inhibits insulin receptor tyrosine kinase activity [1,2,3,4][1][2][3][4]. GSK-3 also inhibits the canonical Wnt signaling pathway through phosphorylation of β-catenin, which de-stabilizes the protein, leading to subsequent degradation in the proteasome [5,6][5][6]. In addition, GSK-3 phosphorylates a variety of transcription factors including Nuclear Factor of Activated T-Cells, NFAT [7], heat shock factor-1 [8], cAMP response element binding protein, CREB, [9], and nuclear factor-kappa B, NF-κB [10], to inhibit gene expression. The unique properties of GSK-3 may explain its involvement in such a wide variety of biological processes: Unlike most of protein kinases, GSK-3 is active under “basal” conditions and is inhibited when cells are stimulated. The substrate recognition is also unusual as it typically requires pre-phosphorylation of the substrate by another “priming kinase” [1,11][1][11]. This unique feature adds additional levels of regulation because the ability of GSK to phosphorylate a substrate is conditionally dependent upon the activation of the priming kinase, and that may be controlled by various factors including cell type and cellular context. It should be noted, however, that unprimed substrates had been reported, such as β-catenin, or presenilin-1, as demonstrated by using a GSK-3 mutant that cannot interact with primed substrates [12,13,14][12][13][14]. Finally, the versatility of GSK-3 also relies on its broad range of substrate, including a predicted number over 500 substrates and about 100 “physiological substrates” that are related to diverse cellular functions [15,16][15][16]. Another important feature of GSK-3 is the existence of two isozymes, GSK-3α and GSK-3β coded by two different genes [17], and a spliced variant of GSK-3β (GSK-3β2) containing a 13 amino acid insert has been described [18]. The GSK-3β2 variant is enriched in neurons and shows lower in vitro activity as compared to GSK-3β [19]. GSK-3 isozymes exhibit both similar and distinct functions. In some cases, the isozymes fulfil non-redundant physiological functions, but in others, there is a possibility of compensation. The GSK-3 isozymes share 97% identity in their catalytic domains, but there are significant differences at the N–and C-terminal domains [17]. Notably, GSK-3α has been largely overlooked in favor of studies with GSK-3β, although roles for GSK-3α in cellular regulation and diseases pathogenesis have recently been described. From the evolutionary perspective, the α and β isozymes split from a common precursor approximately at the time of emergence of vertebrates, and both genes are highly conserved in fish, amphibians, reptiles, and mammals [20]. An interesting finding is that the α gene is missing in birds. Although the initial findings were based on the available draft genome of three species, namely, chickens, domestic turkeys, and zebra-finches [20], searching the updated genomic data confirms the general selective loss of GSK-3α in the avian species (results from our laboratory).
The question of whether or not each GSK-3 isozyme possesses distinct functions has been addressed in many intense studies. One possible cause of the differences between the isozymes could stem from their distinct distribution in the brain, where GSK3α is especially abundant in the hippocampus, cerebral cortex, striatum, and cerebellum, while GSK3β is expressed in nearly all brain regions [21]. Another option could be that the differences are due to their distinct phosphorylation pattern of substrates. Thus, specific deletion of each of the GSK3 isozymes in the brain produced a distinct substrate phosphorylation pattern [19]. For example, phosphorylation of Collapsin response mediated proteins, CRMP2 and CRMP4 at phosphorylation sites Thr 509, Thr 514 and Ser 518 was not detectable in cortex lacking GSK3β but was normal in cortex lacking GSK-3α, and phosphorylation of tau at Thr 231, Thr 235, and Se 396 was predominantly catalyzed by GSK-3β [19], although there may also be redundant activity of the GSK-3 isozymes for other substrates such as β-catenin [22].
In the following section, we summarize the results obtained by genetic manipulations of the GSK-3 isozymes, focusing on phenotypes and processes related to the brain and the nervous system. A significant difference between the isozymes is clearly observed in embryonic development: while loss of GSK-3β is lethal, due to liver degeneration and impaired heart development [23[23][24][25],24,25], GSK-3α null mice are viable [26]. However, it is apparently more complex to distinguish the roles played by the individual GSK-3 isozymes in adult neurons. The brains of GSK-3α null mice are smaller, and the mice exhibit more aggressive behavior, reduced exploratory activity, and reduced social interaction than normal controls [27]. The GSK-3α null mice also have a shortened lifespan that is associated with age-related pathology related to cardiac dysfunction, early onset of sarcopenia, and cellular senescence [28]. Selective loss of GSK-3α in neurons has also been shown to alter neuronal architecture and behavior activity [29]. With respect to pathological conditions, knock-down of GSK3α, but not GSK3β, ameliorated amyloid plaque loads and memory deficits in an Alzheimer’s disease (AD) mouse model [30]. In contrast, manipulation of GSK-3β expression resulted in alterations in neuronal structure, mood behavior, and cognitive functions. Selective loss of GSK-3β in the forebrain pyramidal neurons produced anxiolytic (reduced anxiety) and pro-social effects [31], and loss of GSK-3β but not GSK-3α in GABAergic neurons, reversed gamma oscillation deficits and cognitive dysfunction in an NMDA hypofunction model related to schizophrenia [32]. Another “behavior” study reported that GSK-3β heterozygous mice exhibit reduced exploratory and anxiety behavior [33,34][33][34]. The impact of GSK-3β on neuronal structure was further demonstrated in cortical and hippocampal neurons where selective deletion of GSK-3β reduced dendritic spine stability and attenuated excitatory synaptic transmission [35]. Finally, overexpression of GSK3β reduced brain size in transgenic mice [36].
Conditional deletion of both GSK-3 isozymes further highlighted the prominent role of GSK-3 in regulating brain architecture and behavior skills. Genetic elimination of both GSK-3 isozymes by shRNA reduced axon growth, while localized inhibition of both isozymes at the distal axon resulted in axon elongation [37]. Conditional deletion of GSK-3α and GSK-3β in astrocytes resulted in a larger brain with an increased number of astrocytes [38]. These animals showed aberrant anxiety and altered social behavior [38]. Specific deletion of GSK-3 isozymes in new born cortical neurons, disrupted dendritic orientation and radial migration (moving neurons to a different brain layer) in all areas of the cortex and hippocampus [39]. Finally, deletion of both GSK-3 isozymes in neuronal progenitors resulted in a massive proliferation of cells and prevented progenitor differentiation [40].
The observation that birds lack GSK-3α provides an opportunity to distinguish the specific roles of GSK-3β. Inhibition of brain GSK-3β in a zebra finch model altered singing behavior and reduced neurogenesis in certain regions of the ventricular zone [41]. The results suggested that GSK-3α may be the major tau kinase in the adult brain, as levels of phosphorylated tau (at GSK-3 phosphorylation site) in the bird’s brain were largely reduced as compared to that of found in the mouse brain, a phenomenon that was also recapitulated in the brain of GSK-3α KO mice [20]. As high levels of tau phosphorylation was found in the bird’s embryo, it was further suggested that GSK-3β may be the dominant tau kinase during embryonic development [20]. Interestingly, overexpression of GSK-3β resulted in increased tau phosphorylation in the adult mouse brain [36,42][36][42]. Thus, it seems that GSK-3α may be the preferred tau kinase in adult; nevertheless, GSK-3β may become a “more dominant” tau kinase in pathological conditions [36,42][36][42].
An interesting alternative model for the study of isozyme function is the phosphorylation-resistant GSK-3α/β knock-in mouse [43]. In these mice, GSK-3 could not be inhibited (via serine phosphorylation) by an upstream kinase. The results confirmed a dominant role for GSK-3β (but not GSK-3α) in regulating muscle glycogen synthase [43], as well as in vivo tau phosphorylation by GSK-3 [36]. These mice showed hyperactivity and mania, which recapitulated symptoms of schizophrenia and manic phase in bipolar disorder [44]; in another study, they showed impairment of neuronal precursor cell proliferation [45]. The recent development of isozyme selective GSK-3 inhibitors also provides an opportunity to distinguish differences in function between the two GSK-3 isozymes. The use of BRD0705 to selectively inhibit GSK-3α (IC50 0.066 μM vs. 0.5 μM of α or β isoform respectively [46]), revealed that inhibition of GSK-3α corrects excessive protein synthesis and ameliorates the susceptibility to audiogenic seizures in Fragile X syndrome (FXS) mice [47]. Conversely, inhibition of GSK-3β by the selective inhibitor, BRD3731 (IC50 0.015 μM vs. 0.215 μM of β or α isoform respectively [46]), reversed gamma oscillation and cognitive dysfunction in a mouse model of schizophrenia [32].
2. GSK-3 in Neurodegeneration
GSK-3 is indeed a crucial player in the nervous system, and a significant factor that contributes to disease pathogenesis. Earlier studies revealed lithium salt, a drug approved for treating psychiatric disorders, as a GSK-3 inhibitor [48,49][48][49]. This finding implicated GSK-3 as a central regulator of mood behavior and psychiatric disorders, a notion that has since been supported by numerous studies. The current paradigm suggests that hyperactivity of GSK-3 is a causative factor in progressive neurodegenerative and psychiatric conditions, while inhibition of GSK-3 may be therapeutic. Indeed, hyperactive GSK-3 was found in the AD brain, and overexpression of GSK-3 in vivo induced AD pathology, cognitive deficits, and gliosis in a number of AD mice models [36,50,51,52,53,54,55][36][50][51][52][53][54][55]. Additional studies have reported that alterations in GSK-3 activity (e.g., either excessive activation, or inhibition) influence emotion, mood behavior, sociability skills, and schizophrenia-like behavior [31,33,44,56,57,58,59,60][31][33][44][56][57][58][59][60]. As a corollary, a reduction in GSK-3 activity reverses the severity of a number of diseases. For example, conditional deletion of GSK-3 in the brain of AD transgenic mouse models (mice expressing APP mutant, tau mutant, or double transgene expressing APP/PS1 mutants), was reported to reduce β-amyloid loads and levels of tau phosphorylation, and to decrease the formation of neurofibrillary tangles [30,61][30][61]. Likewise, treatment with GSK-3 inhibitors has been shown to improve disease symptoms in animal models of AD, Parkinson’s disease (PD), amyotrophic lateral sclerosis (ALS), Fragile X syndrome (FXS). Yet, no efficacy was achieved in phase 2 clinical trial for progressive supranuclear palsy (PSP) with the GSK-3 inhibitor tideglusib. Detailed descriptions of these studies have been published elsewhere [62,63,64,65,66,67][62][63][64][65][66][67].
It is evident from the accumulated data that GSK-3 plays a prominent role in regulating structural and metabolic processes both in developing and adult neurons. In this review, we describe the role of GSK-3 in regulating cytoskeleton organization, the mammalian target of rapamycin (mTOR) pathway, and in mitochondria, all of which are components that link GSK-3 to neurodegeneration (see Figure 1). In addition, we provide an update of the field of GSK-3 inhibitors.
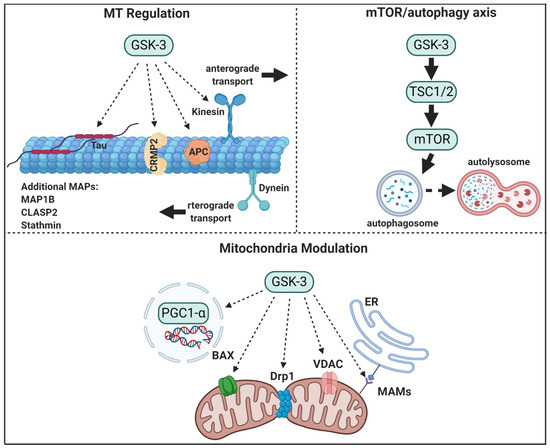
Figure 1. GSK-3 regulatory pathways in neurons. GSK-3 regulates microtubule (MT) stability and dynamics. Phosphorylation of MT binding proteins (MAPs) by GSK-3 reduces their binding to MT, and GSK-3 phosphorylation of kinesin 1 impairs anterograde and retrograde transport. GSK-3 activation of mTORC1 inhibits autophagic and lysosomal activity. GSK3 regulates mitochondrial energy metabolism and mitochondria-mediated cell death. GSK-3 destabilizes peroxisome proliferator-activated receptor γ, PGC1α, and inhibits its transcriptional activity, phosphorylation of dynamin-related protein1, DRP1, by GSK3 enhances mitochondria fission, and phosphorylation of Voltage-dependent anion-selective channel 1, VADC1, and bcl-2 associated proteins, Bax, by GSK-3 enhances their induced-apoptotic activity. Finally, GSK-3 impairs mitochondria and ER communication by disrupting proteins associated with the microdomain, mitochondria-associated membranes, MAM.
References
- Woodgett, J.R.; Cohen, P. Multisite phosphorylation of glycogen synthase. Molecular basis for the substrate specificity of glycogen synthase kinase-3 and casein kinase- II (glycogen synthase kinase-5). Biochim. Biophys. Acta 1984, 788, 339–347.
- Fiol, C.J.; Mahrenholz, A.M.; Wang, Y.; Roeske, R.W.; Roach, P.J. Formation of protein kinase recognition sites by covalent modification of the substrate. Molecular mechanism for the synergistic action of casein kinase II and glycogen synthase kinase 3. J. Biol. Chem. 1987, 262, 14042–14048.
- Liberman, Z.; Eldar-Finkelman, H. Serine 332 phosphorylation of insulin receptor substrate-1 by glycogen synthase kinase-3 attenuates insulin signaling. J. Biol. Chem. 2005, 280, 4422–4428.
- Sharfi, H.; Eldar-Finkelman, H. Sequential phosphorylation of insulin receptor substrate-2 by glycogen synthase kinase-3 and c-Jun NH2-terminal kinase plays a role in hepatic insulin signaling. Am. J. Physiol. Endocrinol. Metab. 2008, 294, E307–E315.
- Yost, C.; Torres, M.; Miller, J.; Huang, E.; Kimelman, D.; Moon, R. The axis-inducing activity, stability, and subcellular distribution of beta-catenin is regulated in Xenopus embryos by glycogen synthase kinase 3. Genes. Dev. 1996, 10, 1443–1454.
- Ikeda, S.; Kishida, S.; Yamamoto, H.; Murai, H.; Koyama, S.; Kikuchi, A. Axin, a negative regulator of the Wnt signaling pathway, forms a complex with GSK-3beta and beta-catenin and promotes GSK-3beta-dependent phosphorylation of beta-catenin. EMBO J. 1998, 17, 1371–1384.
- Beals, C.R.; Sheridan, C.M.; Turck, C.W.; Gardner, P.; Crabtree, G.R. Nuclear export of NF-ATc enhanced by glycogen synthase kinase-3. Science 1997, 275, 1930–1934.
- Chu, B.; Soncin, F.; Price, B.D.; Stevenson, M.A.; Calderwood, S.K. Sequential phosphorylation by mitogen-activated protein kinase and glycogen synthase kinase 3 represses transcriptional activation by heat shock factor-1. J. Biol. Chem. 1996, 271, 30847–30857.
- Grimes, C.A.; Jope, R.S. CREB DNA binding activity is inhibited by glycogen synthase kinase-3 beta and facilitated by lithium. J. Neurochem. 2001, 78, 1219–1232.
- Sanchez, J.F.; Sniderhan, L.F.; Williamson, A.L.; Fan, S.; Chakraborty-Sett, S.; Maggirwar, S.B. Glycogen synthase kinase 3beta-mediated apoptosis of primary cortical astrocytes involves inhibition of nuclear factor kappaB signaling. Mol. Cell. Biol. 2003, 23, 4649–4662.
- Fiol, C.J.; Williams, J.S.; Chou, C.H.; Wang, Q.M.; Roach, P.J.; Andrisani, O.M. A secondary phosphorylation of CREB341 at Ser129 is required for the cAMP-mediated control of gene expression. A role for glycogen synthase kinase-3 in the control of gene expression. J. Biol. Chem. 1994, 269, 32187–32193.
- Twomey, C.; McCarthy, J.V. Presenilin-1 is an unprimed glycogen synthase kinase-3beta substrate. FEBS Lett. 2006, 580, 4015–4020.
- Frame, S.; Cohen, P.; Biondi, R.M. A common phosphate binding site explains the unique substrate specificity of GSK3 and its inactivation by phosphorylation. Mol. Cell 2001, 7, 1321–1327.
- Hagen, T.; Di Daniel, E.; Culbert, A.A.; Reith, A.D. Expression and characterization of GSK-3 mutants and their effect on beta-catenin phosphorylation in intact cells. J. Biol. Chem. 2002, 277, 23330–23335.
- Linding, R.; Jensen, L.J.; Ostheimer, G.J.; van Vugt, M.A.; Jorgensen, C.; Miron, I.M.; Diella, F.; Colwill, K.; Taylor, L.; Elder, K.; et al. Systematic discovery of in vivo phosphorylation networks. Cell 2007, 129, 1415–1426.
- Alabed, Y.Z.; Pool, M.; Ong Tone, S.; Sutherland, C.; Fournier, A.E. GSK3 beta regulates myelin-dependent axon outgrowth inhibition through CRMP4. J. Neurosci. 2011, 30, 5635–5643.
- Woodgett, J.R. Molecular cloning and expression of glycogen synthase kinase-3/factorA. EMBO J. 1990, 9, 2431–2438.
- Mukai, F.; Ishiguro, K.; Sano, Y.; Fujita, S.C. Alternative splicing isoform of tau protein kinase I/glycogen synthase kinase 3beta. J. Neurochem. 2002, 81, 1073–1083.
- Soutar, M.P.; Kim, W.Y.; Williamson, R.; Peggie, M.; Hastie, C.J.; McLauchlan, H.; Snider, W.D.; Gordon-Weeks, P.R.; Sutherland, C. Evidence that glycogen synthase kinase-3 isoforms have distinct substrate preference in the brain. J. Neurochem. 2011, 115, 974–983.
- Alon, L.T.; Pietrokovski, S.; Barkan, S.; Avrahami, L.; Kaidanovich-Beilin, O.; Woodgett, J.R.; Barnea, A.; Eldar-Finkelman, H. Selective loss of glycogen synthase kinase-3alpha in birds reveals distinct roles for GSK-3 isozymes in tau phosphorylation. FEBS Lett. 2011, 585, 1158–1162.
- Yao, H.B.; Shaw, P.C.; Wong, C.C.; Wan, D.C. Expression of glycogen synthase kinase-3 isoforms in mouse tissues and their transcription in the brain. J. Chem. Neuroanat. 2002, 23, 291–297.
- Doble, B.W.; Patel, S.; Wood, G.A.; Kockeritz, L.K.; Woodgett, J.R. Functional redundancy of GSK-3alpha and GSK-3beta in Wnt/beta-catenin signaling shown by using an allelic series of embryonic stem cell lines. Dev. Cell 2007, 12, 957–971.
- Hoeflich, K.P.; Luo, J.; Rubie, E.A.; Tsao, M.S.; Jin, O.; Woodgett, J.R. Requirement for glycogen synthase kinase-3beta in cell survival and NF-kappaB activation. Nature 2000, 406, 86–90.
- Force, T.; Woodgett, J.R. Unique and overlapping functions of GSK-3 isoforms in cell differentiation and proliferation and cardiovascular development. J. Biol. Chem. 2009, 284, 9643–9647.
- Kerkela, R.; Kockeritz, L.; Macaulay, K.; Zhou, J.; Doble, B.W.; Beahm, C.; Greytak, S.; Woulfe, K.; Trivedi, C.M.; Woodgett, J.R.; et al. Deletion of GSK-3beta in mice leads to hypertrophic cardiomyopathy secondary to cardiomyoblast hyperproliferation. J. Clin. Investig. 2008, 118, 3609–3618.
- MacAulay, K.; Doble, B.W.; Patel, S.; Hansotia, T.; Sinclair, E.M.; Drucker, D.J.; Nagy, A.; Woodgett, J.R. Glycogen synthase kinase 3alpha-specific regulation of murine hepatic glycogen metabolism. Cell Metab. 2007, 6, 329–337.
- Kaidanovich-Beilin, O.; Lipina, T.V.; Takao, K.; van Eede, M.; Hattori, S.; Laliberté, C.; Khan, M.; Okamoto, K.; Chambers, J.W.; Fletcher, P.J.; et al. Abnormalities in brain structure and behavior in GSK-3alpha mutant mice. Mol. Brain 2009, 2, 1–23.
- Zhou, J.; Freeman, T.A.; Ahmad, F.; Shang, X.; Mangano, E.; Gao, E.; Farber, J.; Wang, Y.; Ma, X.L.; Woodgett, J.; et al. GSK-3alpha is a central regulator of age-related pathologies in mice. J. Clin. Investig. 2013, 123, 1821–1832.
- Maurin, H.; Lechat, B.; Dewachter, I.; Ris, L.; Louis, J.V.; Borghgraef, P.; Devijver, H.; Jaworski, T.; Van Leuven, F. Neurological characterization of mice deficient in GSK3alpha highlight pleiotropic physiological functions in cognition and pathological activity as Tau kinase. Mol. Brain 2013, 6, 27.
- Hurtado, D.E.; Molina-Porcel, L.; Carroll, J.C.; Macdonald, C.; Aboagye, A.K.; Trojanowski, J.Q.; Lee, V.M. Selectively Silencing GSK-3 Isoforms Reduces Plaques and Tangles in Mouse Models of Alzheimer’s Disease. J. Neurosci. 2012, 32, 7392–7402.
- Latapy, C.; Rioux, V.; Guitton, M.J.; Beaulieu, J.M. Selective deletion of forebrain glycogen synthase kinase 3beta reveals a central role in serotonin-sensitive anxiety and social behaviour. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2012, 367, 2460–2474.
- Nakao, K.; Singh, M.; Sapkota, K.; Hagler, B.C.; Hunter, R.N.; Raman, C.; Hablitz, J.J.; Nakazawa, K. GSK3beta inhibition restores cortical gamma oscillation and cognitive behavior in a mouse model of NMDA receptor hypofunction relevant to schizophrenia. Neuropsychopharmacology 2020, 45, 2207–2218.
- O’Brien, W.T.; Harper, A.D.; Jove, F.; Woodgett, J.R.; Maretto, S.; Piccolo, S.; Klein, P.S. Glycogen synthase kinase-3beta haploinsufficiency mimics the behavioral and molecular effects of lithium. J. Neurosci. 2004, 24, 6791–6798.
- Bersudsky, Y.; Shaldubina, A.; Kozlovsky, N.; Woodgett, J.R.; Agam, G.; Belmaker, R.H. Glycogen synthase kinase-3beta heterozygote knockout mice as a model of findings in postmortem schizophrenia brain or as a model of behaviors mimicking lithium action: Negative results. Behav. Pharmacol. 2008, 19, 217–224.
- Ochs, S.M.; Dorostkar, M.M.; Aramuni, G.; Schon, C.; Filser, S.; Poschl, J.; Kremer, A.; Van Leuven, F.; Ovsepian, S.V.; Herms, J. Loss of neuronal GSK3beta reduces dendritic spine stability and attenuates excitatory synaptic transmission via beta-catenin. Mol. Psychiatry 2015, 20, 482–489.
- Spittaels, K.; Van den Haute, C.; Van Dorpe, J.; Terwel, D.; Vandezande, K.; Lasrado, R.; Bruynseels, K.; Irizarry, M.; Verhoye, M.; Van Lint, J.; et al. Neonatal neuronal overexpression of glycogen synthase kinase-3 beta reduces brain size in transgenic mice. Neuroscience 2002, 113, 797–808.
- Kim, W.Y.; Zhou, F.Q.; Zhou, J.; Yokota, Y.; Wang, Y.M.; Yoshimura, T.; Kaibuchi, K.; Woodgett, J.R.; Anton, E.S.; Snider, W.D. Essential Roles for GSK-3s and GSK-3-Primed Substrates in Neurotrophin-Induced and Hippocampal Axon Growth. Neuron 2006, 52, 981–996.
- Jung, E.M.; Ka, M.; Kim, W.Y. Loss of GSK-3 Causes Abnormal Astrogenesis and Behavior in Mice. Mol. Neurobiol. 2016, 53, 3954–3966.
- Morgan-Smith, M.; Wu, Y.; Zhu, X.; Pringle, J.; Snider, W.D. GSK-3 signaling in developing cortical neurons is essential for radial migration and dendritic orientation. eLife 2014, 3, e02663.
- Kim, W.Y.; Wang, X.; Wu, Y.; Doble, B.W.; Patel, S.; Woodgett, J.R.; Snider, W.D. GSK-3 is a master regulator of neural progenitor homeostasis. Nat. Neurosci. 2009, 12, 1390–1397.
- Aloni, E.; Shapira, M.; Eldar-Finkelman, H.; Barnea, A. GSK-3beta Inhibition Affects Singing Behavior and Neurogenesis in Adult Songbirds. Brain Behav. Evol. 2015, 85, 233–244.
- Lucas, J.J.; Hernandez, F.; Gomez-Ramos, P.; Moran, M.A.; Hen, R.; Avila, J. Decreased nuclear beta-catenin, tau hyperphosphorylation and neurodegeneration in GSK-3beta conditional transgenic mice. EMBO J. 2001, 20, 27–39.
- McManus, E.J.; Sakamoto, K.; Armit, L.J.; Ronaldson, L.; Shpiro, N.; Marquez, R.; Alessi, D.R. Role that phosphorylation of GSK3 plays in insulin and Wnt signalling defined by knockin analysis. EMBO J. 2005, 24, 1571–1583.
- Prickaerts, J.; Moechars, D.; Cryns, K.; Lenaerts, I.; van Craenendonck, H.; Goris, I.; Daneels, G.; Bouwknecht, J.A.; Steckler, T. Transgenic mice overexpressing glycogen synthase kinase 3beta: A putative model of hyperactivity and mania. J. Neurosci. 2006, 26, 9022–9029.
- Eom, T.Y.; Jope, R.S. Blocked inhibitory serine-phosphorylation of glycogen synthase kinase-3alpha/beta impairs in vivo neural precursor cell proliferation. Biol. Psychiatry 2009, 66, 494–502.
- Wagner, F.F.; Benajiba, L.; Campbell, A.J.; Weiwer, M.; Sacher, J.R.; Gale, J.P.; Ross, L.; Puissant, A.; Alexe, G.; Conway, A.; et al. Exploiting an Asp-Glu “switch” in glycogen synthase kinase 3 to design paralog-selective inhibitors for use in acute myeloid leukemia. Sci. Transl. Med. 2018, 10, eaam8460.
- McCamphill, P.K.; Stoppel, L.J.; Senter, R.K.; Lewis, M.C.; Heynen, A.J.; Stoppel, D.C.; Sridhar, V.; Collins, K.A.; Shi, X.; Pan, J.Q.; et al. Selective inhibition of glycogen synthase kinase 3alpha corrects pathophysiology in a mouse model of fragile X syndrome. Sci. Transl. Med. 2020, 12, eaam8572.
- Klein, P.S.; Melton, D.A. A Molecular Mechanism for the Effect of Lithium on Development. Proc. Natl. Acad. Sci. USA 1996, 93, 8455–8459.
- O’Brien, W.T.; Klein, P.S. Validating GSK3 as an in vivo target of lithium action. Biochem. Soc. Trans. 2009, 37 Pt 5, 1133–1138.
- McCamphill, P.K.; Stoppel, L.J.; Senter, R.K.; Lewis, M.C.; Heynen, A.J.; Stoppel, D.C.; Sridhar, V.; Collins, K.A.; Shi, X.; Pan, J.Q.; et al. Glycogen synthase kinase-3 is increased in white cells early in Alzheimer’s disease. Neurosci. Lett. 2005, 373, 1–4.
- Leroy, K.; Yilmaz, Z.; Brion, J.P. Increased level of active GSK-3beta in Alzheimer’s disease and accumulation in argyrophilic grains and in neurones at different stages of neurofibrillary degeneration. Neuropathol. Appl. Neurobiol. 2007, 33, 43–55.
- Pei, J.J.; Tanaka, T.; Tung, Y.C.; Braak, E.; Iqbal, K.; Grundke-Iqbal, I. Distribution, levels, and activity of glycogen synthase kinase-3 in the Alzheimer disease brain. J. Neuropathol. Exp. Neurol. 1997, 56, 70–78.
- Terwel, D.; Muyllaert, D.; Dewachter, I.; Borghgraef, P.; Croes, S.; Devijver, H.; Van Leuven, F. Amyloid activates GSK-3beta to aggravate neuronal tauopathy in bigenic mice. Am. J. Pathol. 2008, 172, 786–798.
- Hernandez, F.; Borrell, J.; Guaza, C.; Avila, J.; Lucas, J.J. Spatial learning deficit in transgenic mice that conditionally over-express GSK-3beta in the brain but do not form tau filaments. J. Neurochem. 2002, 83, 1529–1533.
- Engel, T.; Hernandez, F.; Avila, J.; Lucas, J.J. Full reversal of Alzheimer’s disease-like phenotype in a mouse model with conditional overexpression of glycogen synthase kinase-3. J. Neurosci. 2006, 26, 5083–5090.
- Kaidanovich-Beilin, O.; Milman, A.; Weizman, A.; Pick, C.; Eldar-Finkelman, H. Rapid anti-depressive like activity of specific GSK-3 inhibitor, and its effect on beta-catenin in the mouse hippocampus. Biol. Psychiatry 2004, 55, 781–784.
- Polter, A.; Beurel, E.; Yang, S.; Garner, R.; Song, L.; Miller, C.A.; Sweatt, J.D.; McMahon, L.; Bartolucci, A.A.; Li, X.; et al. Deficiency in the inhibitory serine-phosphorylation of glycogen synthase kinase-3 increases sensitivity to mood disturbances. Neuropsychopharmacology 2010, 35, 1761–1774.
- Mines, M.A.; Yuskaitis, C.J.; King, M.K.; Beurel, E.; Jope, R.S. GSK3 influences social preference and anxiety-related behaviors during social interaction in a mouse model of fragile X syndrome and autism. PLoS ONE 2010, 5, e9706.
- Du, J.; Wei, Y.; Liu, L.; Wang, Y.; Khairova, R.; Blumenthal, R.; Tragon, T.; Hunsberger, J.G.; Machado-Vieira, R.; Drevets, W.; et al. A kinesin signaling complex mediates the ability of GSK-3beta to affect mood-associated behaviors. Proc. Natl. Acad. Sci. USA 2010, 107, 11573–11578.
- Emamian, E.S.; Hall, D.; Birnbaum, M.J.; Karayiorgou, M.; Gogos, J.A. Convergent evidence for impaired AKT1-GSK3beta signaling in schizophrenia. Nat. Genet. 2004, 36, 131–137.
- Ly, P.T.; Wu, Y.; Zou, H.; Wang, R.; Zhou, W.; Kinoshita, A.; Zhang, M.; Yang, Y.; Cai, F.; Woodgett, J.; et al. Inhibition of GSK3beta-mediated BACE1 expression reduces Alzheimer-associated phenotypes. J. Clin. Investig. 2013, 123, 224–235.
- Eldar-Finkelman, H.; Martinez, A. GSK-3 Inhibitors: Preclinical and Clinical Focus on CNS. Front. Mol. Neurosci. 2011, 4, 32.
- Roca, C.; Campillo, N.E. Glycogen synthase kinase 3 (GSK-3) inhibitors: A patent update (2016–2019). Expert Opin. Ther. Pat. 2020, 30, 863–872.
- Palomo, V.; Martinez, A. Glycogen synthase kinase 3 (GSK-3) inhibitors: A patent update (2014–2015). Expert Opin. Ther. Pat. 2017, 27, 657–666.
- King, M.K.; Pardo, M.; Cheng, Y.; Downey, K.; Jope, R.S.; Beurel, E. Glycogen synthase kinase-3 inhibitors: Rescuers of cognitive impairments. Pharmacol. Ther. 2014, 141, 1–12.
- Lovestone, S.; Boada, M.; Dubois, B.; Hull, M.; Rinne, J.O.; Huppertz, H.J.; Calero, M.; Andres, M.V.; Gomez-Carrillo, B.; Leon, T.; et al. A phase II trial of tideglusib in Alzheimer’s disease. J. Alzheimers Dis. 2015, 45, 75–88.
- Tolosa, E.; Litvan, I.; Hoglinger, G.U.; Burn, D.; Lees, A.; Andres, M.V.; Gomez-Carrillo, B.; Leon, T.; Del Ser, T.; Investigators, T. A phase 2 trial of the GSK-3 inhibitor tideglusib in progressive supranuclear palsy. Mov. Disord. 2014, 29, 470–478.