Bacteria live in environments that are in constant flux, and therefore have developed numerous methods to adapt to their ever-changing surroundings. One of these methods of adaptation is called Phase Variation (PV) which is a mechanism of -high-frequency reversible gene expression switching that enables bacteria to generate heterogeneity to successfully compete in uncertain conditions. This entry details the mechanisms of PV and takes a look at them in the context of examples from different bacterial species, with a focus on S. aureus.
- Staphylococcus aureus
- phase variation
1. Introduction
The Gram-positive human commensal Staphylococcus aureus is an opportunistic pathogen that imposes a major health and economic burden on a global scale [1]. S. aureus can colonize multiple sites of the human body, but the primary niche of commensal colonization is the anterior nares with various other skin surfaces making up secondary niches. There are three main carrier-patterns of S. aureus amongst healthy individuals: persistent carriers (~20%), intermittent carriers (~30%), and non-carriers (~50%) [2] [2] and nasal carriage has been linked to a higher chance of contracting infection [2]. S. aureus is responsible for an astounding diversity of infections including infective endocarditis, osteoarticular infections, surgical site infections, and bacteraemia [3,4][3][4]. S. aureus can also cause pneumonia and other respiratory infections, particularly in people living with cystic fibrosis [3]. Furthermore, S. aureus is supremely adept at colonizing alien surfaces within the body and is often responsible for infections associated with catheters, cannula, artificial heart valves, and prosthetic joints [3]. This diverse range of infections is enabled by a vast arsenal of virulence factors that are ready to be deployed in a variety of host environments [5,6][5][6]. Additionally, and of growing concern is S. aureus’ ability to rapidly develop antibiotic resistance. Methicillin Resistant S. aureus (MRSA) has broad-spectrum resistance against the β-lactam group of antibiotics and is a global danger with clones existing in both nosocomial and community settings [7]. MRSA is also a problem in the livestock sector, where it can co-infect both animals and humans [8]. The infamous development of antibiotic resistance, coupled with its worrying genetic plasticity, has earned S. aureus a place in the ESKAPE group of pathogens: a collection of bacteria that represent paradigms of acquisition, development, and transfer of antibiotic resistance [9]. Thus, to better combat this dangerous pathogen it is vitally important to study adaptation mechanisms of S. aureus.
An intriguing trait of S. aureus that makes it notoriously difficult to combat in the clinical setting is phenotypic heterogeneity. An example of this is the phenomenon of persister cells, where sub-populations of S. aureus gain a resistance phenotype against antibiotic treatment resulting from arrested growth [10]. Persister cells may be generated in numerous ways, one of which is the formation of Small Colony Variants (SCVs) that are characterized by auxotrophy for various compounds involved in the electron transport chain and slow growth, allowing them to escape the effects of many antibiotics [11,12][11][12]. Importantly, these populations do not acquire conventional resistance mechanisms against the antibiotics. This heterogenous phenomenon has severe clinical implications and is thought to be a significant cause of antibiotic treatment failure and chronic recurrent infections [13].
Heterogeneity is not, however, limited to antibiotic resistance and diverse traits have been recognised as being expressed in sub-populations. Phase Variation (PV) is among the methods bacteria can employ to generate heterogeneity. PV is a mechanism of high-frequency reversible gene switching that allows sub-populations of bacteria to switch gene expression ON or OFF. While focus is increasingly shifting towards the investigation of such phenomena, more work must be done to fully elucidate the various mechanisms employed by S. aureus to generate heterogeneity and improve it's adaptability to its ever-changing environment. The following entry discusses Phase Variation (PV) and its mechanisms in the context of bacterial adaptation (with a focus on S. aureus) to fluctuating environments.
2. Bacterial Phase Variation
2.1. Background of Phase Variation
All living organisms are faced with the constant challenge of maintaining fitness in order to survive and reproduce, and this is no less true for bacterial species. Bacteria are under constant onslaught from fluctuations in their local environment, infection from bacteriophages, and (in the case of pathogenic bacteria) attack from their infected host. Although bacteria possess robust mechanisms of classical gene regulation that allow them to respond to extracellular changes (e.g., Bacterial Two-Component Systems), these alone may be unable to cope with the constant barrage of fluctuating pressures they face. These selective pressures are often focused on bacterial external proteins which form the first line of contact with the outside environment and this has led to development of what have been termed “contingency loci” [14,15][14][15]. Contingency loci are hypermutable genes that generate genetic and phenotypic variation allowing bacterial populations to survive unpredictable pressures. This hypermutability is conferred by the phenomena of Phase Variation (PV) and antigenic variation.
PV is a reversible gene expression switch that can alter expression between an ON and an OFF state and occurs through several genetic and epigenetic mechanisms [16]. It is characterized by high frequencies, usually exceeding 1 × 10−5 variants per total number of cells [17][18] [17,18] which is orders of magnitude above the typical frequencies of spontaneous mutations (10−6 to 10−8 per cell per generation) [18]. Depending on the method of calculation, the frequency of PV may describe not only rate of the PV mechanisms but also the growth of the phase variants themselves. Antigenic variation is related to PV and occurs through similar mechanisms. However, rather than alternating between an ON and OFF state, antigenic variation mechanisms generate variations in the sequence of surface proteins resulting in the expression of different forms and structures of the antigenic proteins on the cell surface [17,18,19][17][18][19].
As mentioned above, genes subject to PV often encode for cell-surface associated features such as adhesins, liposaccharide synthesis enzymes, and pili [20,21,22][20][21][22] but can also encode for virulence factors and secreted proteins such as iron acquisition machinery [23,24][23][24]. The collection of phase variable loci in a bacterial species is referred to as the “phasome” [16] [16] and generally includes genes which are involved in bottlenecks experienced by the bacterial population. This is most clearly seen in pathogenic bacteria which undergo constant challenge from host immunity during the infection process. For example, PV mediated shutdown of liposaccharide synthesis genes in the invasive pathogen Haemophilus influenzae confers protection against neutrophil-mediated immune clearance but is detrimental in other environments [22,25,26][22][25][26]. In another example, PV in Salmonella typhimurium flagellae can modulate their antigenic properties and allow for evasion from host immunity [27].
It is likely that the original role of PV was as a mechanism of innate immunity against bacteria’s greatest enemy: bacteriophages [28]. Although bacteriophages exist in exaggerated abundance relative to their bacterial hosts, their host range is often limited to just a few specific strains of a given bacterial species [29]. Thus, there is a constant cyclical arms race between bacteria and bacteriophages in order to stay one step ahead of each other [30], and PV plays an important role in both sides of this war. An example can once again be found in liposaccharide synthesis genes of H. influenzae in which PV can result in a switch from a sensitive to resistant phenotype against the HP1c1 phage [31] [31]. On the other hand, PV in the Escherichia coli phage Mu causes a switch in expression between two sets of tail fibers resulting in modulation of the host specificity [32][33] [32,33] with similar phenomena identified in other phages [34].
Considering the above information, it can be inferred that genetic loci susceptible to PV would be found in abundance amongst bacterial species that experience population bottlenecks. Typically, such bottlenecks often occur during the infectious process which imposes limits onto the bacterial population size. These bottlenecks reduce genetic diversity at a time when variation is most beneficial, and PV offers a solution to this hurdle and indeed, several pathogenic bacteria have been documented to undergo PV [18].
While PV is, by definition, a stochastic process, it occurs through several discrete mechanisms. Broadly speaking, mechanisms of generating PV can be discriminated into genetic and epigenetic mechanisms [16] [16] both of which will be addressed in Section 2.2 and Section 2.3 respectively.
2.2. Genetic Mechanisms of PV
There are three genetic mechanisms of PV which shall be discussed in the following chapters: Variation in length of DNA Short Sequence Repeats (SSRs) [35[35][36][37],36,37], DNA inversion [38], and DNA recombination [39,40][39][40].
2.2.1. Variation in Length of DNA Short Sequence Repeats (SSRs)
SSRs are homo- or hetero-nucleotide repeats in DNA that are highly prone to insertion/deletion (indel) errors due to Slipped-Strand Mispairings (SSMs) during DNA replication [35,36,37][35][36][37]. SSRs can be as complex as repeating units of tetranucleotides or as simple as a straight homonucleotide run. Indels in SSRs can result in frameshifts that largely have an ON↔OFF effect on protein function or gene expression (by resulting in abrupt termination of translation or inhibition of RNA polymerase binding, respectively Figure 1A) but can have an alternative gradation effect on gene expression as well. For example, alterations in the length of a dinucleotide TA10 tract in the promoter regions of the divergently transcribed hifA and hifB genes controlling fimbriae expression in H. influenzae can either significantly affect hif expression (TA10→TA9) or only moderately affect it (TA10→TA11) [41]. The evolution of the mutability of SSR tracts is largely driven by a combination of environmental and molecular drivers. The environmental drivers include factors such as the aforementioned population bottlenecks arising during infection processes. These bottleneck conditions exert a primary selective pressure for phenotypes that can survive them, e.g., a population that can shut down the expression of a surface protein that is targeted by host immunity. The necessity to survive this recurrent primary selection serves as a secondary layer of selection for plasticity of the gene itself.
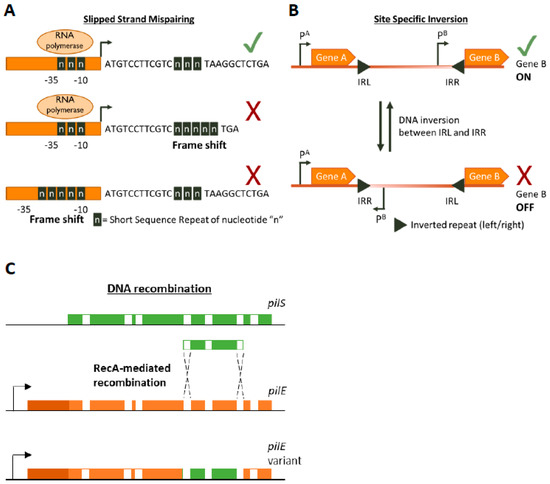
Figure 1. Genetic mechanisms of Phase Variation. A cartoon depicting the three main genetic mechanisms of Phase variation (PV). (A) Slipped-Strand Mispairing events within Short Sequence Repeats (SSR) result in expression (green tick mark) of truncated dysfunctional proteins (if SSR is in the CDS) or inhibition (red cross) of transcription by preventing RNA polymerase/transcription factor binding or by other mechanisms. For example, an interesting method of PV-mediated transcriptional control is shown by Danne et al. who demonstrate SSR alterations upstream of the pilA locus of Streptococcus gallolyticus can destabilize a premature transcription-terminating stem loop [61]. (B) Site-specific inversion is carried out by recombinases that recognize inverted repeat regions (Inverted Repeat Left/Right IRL/IRR) and flip the DNA sequence in between them. If a promoter region (e.g., pB) lies within the sequence flanked by the inverted repeats this leads to shut down of gene expression. (C) RecA-mediated DNA recombination of N. gonorrhoea pilS into pilE results in the formation of new pilE variants. Both pilS and pilE contain variable regions (depicted in green and orange, respectively) interspersed with conserved regions (white) while pilE has a further 5’ conserved region (dark orange) and a promoter to initiate transcription.
The molecular factors are intrinsic to SSR tracts and include the DNA replication and the Mismatch Repair (MMR) [42]. The discriminating factors of SSRs can be broadly delineated into two groups: the composition of the repeating nucleotide unit (i.e., a homonucleotide or a heteronucleotide repeat) and the tract length. These in turn are differentially affected by the DNA replication and MMR machinery. Amongst these proteins are the DNA polymerase enzymes which include the polymerase responsible for the construction of new DNA strands (DNA polymerase III) as well as the polymerase responsible for DNA repair (DNA polymerase I). Studies have shown that these polymerases have an inherent frequency of generating addition/deletion errors when constructing new DNA strands [43,44][43][42]. Following DNA replication, any errors are corrected by the MMR machinery which is a suite of Mut proteins that target and fix errors in a strand specific manner. Inactivation of components from either of these suites of proteins results in a hypermutable phenotype and can lead to SSR alteration e.g., [45]. Additionally, the hypermutable phenotype that results from loss of the MMR machinery is directly responsible for genetic variability of bacteria and mutator phenotypes play an important role in bacterial adaptation [46]. For example, both S. aureus and Pseudomonas aeruginosa isolated from the lungs of people suffering from cystic fibrosis are commonly associated with antibiotic resistance caused by hypermutability [47,48,49][47][48][49]. Interestingly, while both the MMR machinery and DNA polymerases are involved in SSR evolution, they do not appear to be fully redundant. Several studies have shown that MMR is more responsible for variability of homonucleotide SSRs, especially for those which exceed eight nucleotides in length, whereas DNA polymerase I is exclusively responsible for mutations in heteronucleotide SSRs [50,51,52][50][51][52]. This could have evolutionary implications for the mechanisms of generating SSRs. For example, H.influenza is enriched with tetra-nucleotide SSRs [51] [51] whose expansion/contraction is affected by DNA polymerase I. Furthermore, evidence suggests that the frequency of DNA polymerase I mediated errors differs between the leading and the lagging strands of newly synthesized DNA, implying that the direction of genes in the chromosome can also dictate the type of SSR that would evolve in them [53]. Lastly, an interesting study carried out by Lin et al. investigated the distribution of SSRs within the genomes of several bacterial species. They found that in many pathogenic species, SSRs were enriched towards the N-termini of protein coding sequences increasing the probability of frameshifts resulting in non-functional proteins [54,55][54][55]. This further suggests that bacteria have evolved SSRs in a manner to provide maximal PV.
2.2.2. DNA Inversion
DNA inversion was the first documented example of PV, though the mechanism was not known at the time the phenomenon was documented [38] [38] (Figure 1B). It involves recognition of inverted repeat (IR) sequences by invertase enzymes and subsequent enzyme-mediated inversion of the DNA. An elaborate study was carried out by Jiang and colleagues who developed an algorithm to search published bacterial genome datasets for IR sequences that might be phase variable [56]. Not only did they identify that IR sequences were enriched in host-associates species (implying a benefit of PV during commensalism or infection) but they also discovered three antibiotic resistance genes regulated by invertible promoters: a macrolide resistance gene, a multidrug resistance cassette conferring resistance to macrolides and cephalosporins, and a cationic antibacterial peptide resistance operon [56]. The presence of antibiotics influenced the switch from an OFF to an ON state for these genes. Some of the invertible promoters seem to be located on genetic elements homologous to those conveyable by horizontal gene transfer mechanisms, raising the worrying possibility that these resistance gene switches can be transferred to other species [56].
2.2.3. DNA Recombination
Homologous recombination provides a pathway for DNA re-arrangement and subsequent PV. Events arising from recombination mechanisms are often due to DNA deletions, and thus tend to be in a one way ON→OFF direction. However, gene duplication or transfer events can often occur to balance out the accumulation of inactive variants in the population. A well characterized example of recombination mediated variation occurs in the Neisseria gonorrhoea pilus organelle, which is essential for full infectivity and natural transformation. N. gonorrhoea contains a pilE gene that encodes for a pilin protein that is the major component of the pilus, but also contains several silent pilS alleles several Kb away [39]. RecA-dependent recombination events can unidirectionally transfer large sections of the pilS allele into pilE, thus creating an OFF variant [40][57] [40,57] (Figure 1C). The N. gonorrhoea pilus also undergoes PV by SSM-mediated variation in the length of a poly(G) tract in the pilC gene (which encodes for the adhesive tip of the pilus [58]) resulting in ON↔OFF switching [59,60][59][60].
2.3. Epigenetic Mechanisms of Phase Variation
An epigenetic trait has been defined as a heritable phenotype resulting from modified gene expression that is not due to any alterations in the DNA sequence of the chromosome [62,63][62][63]. In prokaryotes, DNA methylation occurs mainly at the nucleotide adenine although studies have shown that cytosine methylation can also occur [64,65,66][64][65][66]. DNA methylation usually occurs at specific target sites and is carried out either by methyltransferases that are part of dedicated Restriction–Modification (RM) systems or by orphan methyltransferases. A well-studied methylase responsible for bacterial epigenetic regulation of PV is the DNA Adenine Methyltransferase (DAM) which is an orphan methyltransferase of the gammaproteobacterial family that is specific for GATC sites [64]. Methylation of DNA represses transcription, and thus PV can result if there are GATC sites within a gene promoter which also binds transcription factors, causing mutually exclusive binding competition between the transcription factor(s) and DAM. If there are numerous GATC sites within a promoter region then the mutually exclusive competition can result in differential methylation patterns of the promoter region resulting in switching between an ON and OFF state. A paradigm of this sort of PV is established by a series of intriguing reports studying the pap operon of E. coli and the opvAB operon of Salmonella enterica [21,67,68,69][21][67][68][69].
2.4. Combined Mechanisms of Phase Variation
There is growing evidence that shows that many bacterial species undertake a combined approach for PV to maximize the ability to generate rapid and diverse variation. This strategy involves generating PV through genetic mechanisms in genes of RM systems that can modify the transcriptome of the cell via epigenetic control. Such systems are referred to as “phasevarions” as they control phase-variable regulons [70] [70] and are immensely powerful weapons in the arsenal of pathogens.
The earliest phasevarions identified are controlled by Type III RM systems. PV occurs in SSRs in the mod gene resulting in ON↔OFF variation and altered methylation states [71,72][71][72]. Strikingly, analyses of known Type III system sequences indicate that at least 20% of these systems contain SSRs and could potentially be phasevarions [73]. Furthermore, mod genes are highly conserved, with variation occurring mainly in the DNA recognition domain. This allows mod genes to exist within the species as multiple alleles, each of which controls distinct phasevarions [16].
There is some evidence of a Type II RM regulated phasevarion detected in Campylobacter jejuni, and gene expression patterns were detectably different upon RNAseq analysis, though no direct link to any altered phenotype was reported [74].
PV in Type I systems largely occurs through DNA inversion in the hsdS gene, creating multiple allelic variants of the specificity protein of the Type I system resulting in different gene targets upon PV [75]. An example of a Type I RM phasevarions can be seen in variable capsular expression controlling virulence in Streptococcus pneumonia [16,76][16][76].
References
- Cosgrove, S.E.; Qi, Y.; Kaye, K.S.; Harbarth, S.; Karchmer, A.W.; Carmeli, Y. The impact of methicillin resistance in Staphylococcus aureus bacteremia on patient outcomes: Mortality, length of stay, and hospital charges. Infect. Control Hosp. Epidemiol. 2005, 26, 166–174, doi:10.1086/502522.
- Wertheim, H.F.L.; Melles, D.C.; Vos, M.C.; van Leeuwen, W.; van Belkum, A.; Verbrugh, H.A.; Nouwen, J.L. The role of nasal carriage in Staphylococcus aureus infections. Lancet Infect. Dis. 2005, 5, 751–762, doi:10.1016/S1473-3099(05)70295-4.
- Tong, S.Y.; Davis, J.S.; Eichenberger, E.; Holland, T.L.; Fowler, V.G., Jr. Staphylococcus aureus infections: Epidemiology, pathophysiology, clinical manifestations, and management. Clin. Microbiol. Rev. 2015, 28, 603–661, doi:10.1128/cmr.00134-14.
- Reddy, P.N.; Srirama, K.; Dirisala, V.R. An Update on Clinical Burden, Diagnostic Tools, and Therapeutic Options of Staphylococcus aureus. Infect. Dis. Res. Treat. 2017, 10, 1179916117703999, doi:10.1177/1179916117703999.
- Oliveira, D.; Borges, A.; Simões, M. Staphylococcus aureus Toxins and Their Molecular Activity in Infectious Diseases. Toxins 2018, 10, 252, doi:10.3390/toxins10060252.
- Tam, K.; Torres, V.J. Staphylococcus aureus Secreted Toxins and Extracellular Enzymes. Microbiol. Spectr. 2019, 7, doi:10.1128/microbiolspec.GPP3-0039-2018.
- Gajdács, M. The Continuing Threat of Methicillin-Resistant Staphylococcus aureus. Antibiotics 2019, 8, 52.
- Cuny, C.; Wieler, L.H.; Witte, W. Livestock-Associated MRSA: The Impact on Humans. Antibiotics 2015, 4, 521–543.
- Rice, L.B. Federal Funding for the Study of Antimicrobial Resistance in Nosocomial Pathogens: No ESKAPE. J. Infect. Dis. 2008, 197, 1079–1081, doi:10.1086/533452.
- Chang, J.; Lee, R.-E.; Lee, W. A pursuit of Staphylococcus aureus continues: A role of persister cells. Arch. Pharmacal. Res. 2020, 43, 630–638, doi:10.1007/s12272-020-01246-x.
- Garcia, L.G.; Lemaire, S.; Kahl, B.C.; Becker, K.; Proctor, R.A.; Denis, O.; Tulkens, P.M.; Van Bambeke, F. Antibiotic activity against small-colony variants of Staphylococcus aureus: Review of in vitro, animal and clinical data. J. Antimicrob. Chemother. 2013, 68, 1455–1464, doi:10.1093/jac/dkt072.
- Kahl, B.C. Small colony variants (SCVs) of Staphylococcus aureus—A bacterial survival strategy. Infect. Genet. Evol. 2014, 21, 515–522, doi:10.1016/j.meegid.2013.05.016.
- Fauvart, M.; De Groote, V.N.; Michiels, J. Role of persister cells in chronic infections: Clinical relevance and perspectives on anti-persister therapies. J. Med. Microbiol. 2011, 60, 699–709, doi:10.1099/jmm.0.030932-0.
- Moxon, R.; Bayliss, C.; Hood, D. Bacterial Contingency Loci: The Role of Simple Sequence DNA Repeats in Bacterial Adaptation. Annu. Rev. Genet. 2006, 40, 307–333, doi:10.1146/annurev.genet.40.110405.090442.
- Moxon, E.R.; Rainey, P.B.; Nowak, M.A.; Lenski, R.E. Adaptive evolution of highly mutable loci in pathogenic bacteria. Curr. Biol. 1994, 4, 24–33, doi:10.1016/S0960-9822(00)00005-1.
- Phillips, Z.N.; Tram, G.; Seib, K.L.; Atack, J.M. Phase-variable bacterial loci: How bacteria gamble to maximise fitness in changing environments. Biochem. Soc. Trans. 2019, 47, 1131–1141, doi:10.1042/bst20180633.
- Bayliss, C.D. Determinants of phase variation rate and the fitness implications of differing rates for bacterial pathogens and commensals. FEMS Microbiol. Rev. 2009, 33, 504–520, doi:10.1111/j.1574-6976.2009.00162.x.
- Wisniewski-Dyé, F.; Vial, L. Phase and antigenic variation mediated by genome modifications. Antonie Leeuwenhoek 2008, 94, 493–515, doi:10.1007/s10482-008-9267-6.
- Van der Woude, M.W.; Baumler, A.J. Phase and antigenic variation in bacteria. Clin. Microbiol Rev. 2004, 17, 581–611, doi:10.1128/cmr.17.3.581-611.2004.
- Atack, J.M.; Winter, L.E.; Jurcisek, J.A.; Bakaletz, L.O.; Barenkamp, S.J.; Jennings, M.P. Selection and Counterselection of Hia Expression Reveals a Key Role for Phase-Variable Expression of Hia in Infection Caused by Nontypeable Haemophilus influenzae. J. Infect. Dis. 2015, 212, 645–653, doi:10.1093/infdis/jiv103.
- Blyn, L.B.; Braaten, B.A.; Low, D.A. Regulation of pap pilin phase variation by a mechanism involving differential dam methylation states. EMBO J. 1990, 9, 4045–4054, doi:10.1002/j.1460-2075.1990.tb07626.x.
- Fox, K.L.; Atack, J.M.; Srikhanta, Y.N.; Eckert, A.; Novotny, L.A.; Bakaletz, L.O.; Jennings, M.P. Selection for phase variation of LOS biosynthetic genes frequently occurs in progression of non-typeable Haemophilus influenzae infection from the nasopharynx to the middle ear of human patients. PLoS ONE 2014, 9, e90505, doi:10.1371/journal.pone.0090505.
- Richardson, A.R.; Stojiljkovic, I. HmbR, a Hemoglobin-Binding Outer Membrane Protein of Neisseria meningitidis, Undergoes Phase Variation. J. Bacteriol. 1999, 181, 2067–2074, doi:10.1128/jb.181.7.2067-2074.1999.
- Ren, Z.; Jin, H.; Whitby, P.W.; Morton, D.J.; Stull, T.L. Role of CCAA Nucleotide Repeats in Regulation of Hemoglobin and Hemoglobin-Haptoglobin Binding Protein Genes of Haemophilus influenzae. J. Bacteriol. 1999, 181, 5865–5870, doi:10.1128/jb.181.18.5865-5870.1999.
- Fox, K.L.; Yildirim, H.H.; Deadman, M.E.; Schweda, E.K.H.; Moxon, E.R.; Hood, D.W. Novel lipopolysaccharide biosynthetic genes containing tetranucleotide repeats in Haemophilus influenzae, identification of a gene for adding O-acetyl groups. Mol. Microbiol. 2005, 58, 207–216, doi:10.1111/j.1365-2958.2005.04814.x.
- Langereis, J.D.; Weiser, J.N. Shielding of a Lipooligosaccharide IgM Epitope Allows Evasion of Neutrophil-Mediated Killing of an Invasive Strain of Nontypeable Haemophilus influenzae. mBio 2014, 5, e01478-14, doi:10.1128/mBio.01478-14.
- Ikeda, J.S.; Schmitt, C.K.; Darnell, S.C.; Watson, P.R.; Bispham, J.; Wallis, T.S.; Weinstein, D.L.; Metcalf, E.S.; Adams, P.; O’Connor, C.D.; et al. Flagellar Phase Variation of Salmonella enterica Serovar Typhimurium Contributes to Virulence in the Murine Typhoid Infection Model but Does Not Influence Salmonella-Induced Enteropathogenesis. Infect. Immun. 2001, 69, 3021–3030, doi:10.1128/iai.69.5.3021-3030.2001.
- Bikard, D.; Marraffini, L.A. Innate and adaptive immunity in bacteria: Mechanisms of programmed genetic variation to fight bacteriophages. Curr. Opin. Immunol. 2012, 24, 15–20, doi:10.1016/j.coi.2011.10.005.
- Hyman, P.; Abedon, S.T. Chapter 7—Bacteriophage Host Range and Bacterial Resistance. In Advances in Applied Microbiology; Academic Press, 2010; Volume 70, pp. 217–248.
- Stern, A.; Sorek, R. The phage-host arms race: Shaping the evolution of microbes. Bioessays News Rev. Mol. Cell. Dev. Biol. 2011, 33, 43–51, doi:10.1002/bies.201000071.
- Zaleski, P.; Wojciechowski, M.; Piekarowicz, A. The role of Dam methylation in phase variation of Haemophilus influenzae genes involved in defence against phage infection. Microbiology 2005, 151, 3361–3369, doi:10.1099/mic.0.28184-0.
- Kamp, D.; Kahmann, R.; Zipser, D.; Broker, T.R.; Chow, L.T. Inversion of the G DNA segment of phage Mu controls phage infectivity. Nature 1978, 271, 577–580, doi:10.1038/271577a0.
- Van de Putte, P.; Cramer, S.; Giphart-Gassler, M. Invertible DNA determines host specificity of bacteriophage Mu. Nature 1980, 286, 218–222, doi:10.1038/286218a0.
- Sandmeler, H. Acquisition and rearrangement of sequence motifs in the evolution of bacteriophage tail fibres. Mol. Microbiol. 1994, 12, 343–350, doi:10.1111/j.1365-2958.1994.tb01023.x.
- Levinson, G.; Gutman, G.A. Slipped-strand mispairing: A major mechanism for DNA sequence evolution. Mol. Biol. Evol. 1987, 4, 203–221, doi:10.1093/oxfordjournals.molbev.a040442.
- Streisinger, G.; Okada, Y.; Emrich, J.; Newton, J.; Tsugita, A.; Terzaghi, E.; Inouye, M. Frameshift mutations and the genetic code. Cold Spring Harb. Symp. Quant. Biol. 1966, 31, 77–84, doi:10.1101/sqb.1966.031.01.014.
- Streisinger, G.; Owen, J.E. Mechanisms of spontaneous and induced frameshift mutation in bacteriophage T4. Genetics 1985, 109, 633–659.
- Andrewes, F.W. Studies in group-agglutination I. The salmonella group and its antigenic structure. J. Pathol. Bacteriol. 1922, 25, 505–521, doi:10.1002/path.1700250411.
- Sechman, E.V.; Rohrer, M.S.; Seifert, H.S. A genetic screen identifies genes and sites involved in pilin antigenic variation in Neisseria gonorrhoeae. Mol. Microbiol. 2005, 57, 468–483, doi:10.1111/j.1365-2958.2005.04657.x.
- Mehr, I.J.; Seifert, H.S. Differential roles of homologous recombination pathways in Neisseria gonorrhoeae pilin antigenic variation, DNA transformation and DNA repair. Mol. Microbiol. 1998, 30, 697–710, doi:10.1046/j.1365-2958.1998.01089.x.
- Van Ham, S.M.; van Alphen, L.; Mooi, F.R.; van Putten, J.P.M. Phase variation of H. influenzae fimbriae: Transcriptional control of two divergent genes through a variable combined promoter region. Cell 1993, 73, 1187–1196, doi:10.1016/0092-8674(93)90647-9.
- Bayliss, C.D.; Palmer, M.E. Evolution of simple sequence repeat–mediated phase variation in bacterial genomes. Ann. N. Y. Acad. Sci. 2012, 1267, 39–44, doi:10.1111/j.1749-6632.2012.06584.x.
- Thomas, A.; Kunkel, K.B. DNA Replication Fidelity. Annu. Rev. Biochem. 2000, 69, 497–529, doi:10.1146/annurev.biochem.69.1.497.
- Pham, P.T.; Olson, M.W.; McHenry, C.S.; Schaaper, R.M. The Base Substitution and Frameshift Fidelity of Escherichia coli DNA Polymerase III Holoenzyme in Vitro. J. Biol. Chem. 1998, 273, 23575–23584, doi:10.1074/jbc.273.36.23575.
- Richardson, A.R.; Stojiljkovic, I. Mismatch repair and the regulation of phase variation in Neisseria meningitidis. Mol. Microbiol. 2001, 40, 645–655, doi:10.1046/j.1365-2958.2001.02408.x.
- Taddei, F.; Radman, M.; Maynard-Smith, J.; Toupance, B.; Gouyon, P.H.; Godelle, B. Role of mutator alleles in adaptive evolution. Nature 1997, 387, 700–702, doi:10.1038/42696.
- Prunier, A.-L.; Malbruny, B.; Laurans, M.; Brouard, J.; Duhamel, J.-F.; Leclercq, R. High Rate of Macrolide Resistance in Staphylococcus aureus Strains from Patients with Cystic Fibrosis Reveals High Proportions of Hypermutable Strains. J. Infect. Dis. 2003, 187, 1709–1716, doi:10.1086/374937.
- Prunier, A.-L.; Leclercq, R. Role of mutS and mutL Genes in Hypermutability and Recombination in Staphylococcus aureus. J. Bacteriol. 2005, 187, 3455–3464, doi:10.1128/jb.187.10.3455-3464.2005.
- Behzadi, P.; Baráth, Z.; Gajdács, M. It’s Not Easy Being Green: A Narrative Review on the Microbiology, Virulence and Therapeutic Prospects of Multidrug-Resistant Pseudomonas aeruginosa. Antibiotics 2021, 10, 42.
- Martin, P.; Sun, L.; Hood, D.W.; Moxon, E.R. Involvement of genes of genome maintenance in the regulation of phase variation frequencies in Neisseria meningitidis. Microbiology 2004, 150, 3001–3012, doi:10.1099/mic.0.27182-0.
- Bayliss, C.D.; van de Ven, T.; Moxon, E.R. Mutations in poll but not mutSLH destabilize Haemophilus influenzae tetranucleotide repeats. EMBO J. 2002, 21, 1465–1476, doi:10.1093/emboj/21.6.1465.
- Tran, H.T.; Keen, J.D.; Kricker, M.; Resnick, M.A.; Gordenin, D.A. Hypermutability of homonucleotide runs in mismatch repair and DNA polymerase proofreading yeast mutants. Mol. Cell. Biol. 1997, 17, 2859–2865, doi:10.1128/mcb.17.5.2859.
- Gawel, D.; Jonczyk, P.; Bialoskorska, M.; Schaaper, R.M.; Fijalkowska, I.J. Asymmetry of frameshift mutagenesis during leading and lagging-strand replication in Escherichia coli. Mutat. Res./Fundam. Mol. Mech. Mutagenesis 2002, 501, 129–136, doi:10.1016/S0027-5107(02)00020-9.
- Orsi, R.H.; Bowen, B.M.; Wiedmann, M. Homopolymeric tracts represent a general regulatory mechanism in prokaryotes. BMC Genom. 2010, 11, 102, doi:10.1186/1471-2164-11-102.
- Lin, W.-H.; Kussell, E. Evolutionary pressures on simple sequence repeats in prokaryotic coding regions. Nucleic Acids Res. 2011, 40, 2399–2413, doi:10.1093/nar/gkr1078.
- Jiang, X.; Hall, A.B.; Arthur, T.D.; Plichta, D.R.; Covington, C.T.; Poyet, M.; Crothers, J.; Moses, P.L.; Tolonen, A.C.; Vlamakis, H.; et al. Invertible promoters mediate bacterial phase variation, antibiotic resistance, and host adaptation in the gut. Science 2019, 363, 181–187, doi:10.1126/science.aau5238.
- Henderson, I.R.; Owen, P.; Nataro, J.P. Molecular switches—The ON and OFF of bacterial phase variation. Mol. Microbiol. 1999, 33, 919–932.
- Rudel, T.; Scheuerpflug, I.; Meyer, T.F. Neisseria PilC protein identified as type-4 pilus tip-located adhesin. Nature 1995, 373, 357–359, doi:10.1038/373357a0.
- Rytkönen, A.; Albiger, B.; Hansson-Palo, P.; Källström, H.; Olcén, P.; Fredlund, H.; Jonsson, A.-B. Neisseria meningitidis Undergoes PilC Phase Variation and PilE Sequence Variation during Invasive Disease. J. Infect. Dis. 2004, 189, 402–409, doi:10.1086/381271.
- Jonsson, A.B.; Nyberg, G.; Normark, S. Phase variation of gonococcal pili by frameshift mutation in pilC, a novel gene for pilus assembly. EMBO J. 1991, 10, 477–488, doi:10.1002/j.1460-2075.1991.tb07970.x.
- Danne, C.; Dubrac, S.; Trieu-Cuot, P.; Dramsi, S. Single Cell Stochastic Regulation of Pilus Phase Variation by an Attenuation-like Mechanism. PLoS Pathog. 2014, 10, e1003860, doi:10.1371/journal.ppat.1003860.
- Berger, S.L.; Kouzarides, T.; Shiekhattar, R.; Shilatifard, A. An operational definition of epigenetics. Genes Dev. 2009, 23, 781–783, doi:10.1101/gad.1787609.
- Dupont, C.; Armant, D.R.; Brenner, C.A. Epigenetics: Definition, mechanisms and clinical perspective. Semin. Reprod. Med. 2009, 27, 351–357, doi:10.1055/s-0029-1237423.
- Blow, M.J.; Clark, T.A.; Daum, C.G.; Deutschbauer, A.M.; Fomenkov, A.; Fries, R.; Froula, J.; Kang, D.D.; Malmstrom, R.R.; Morgan, R.D.; et al. The Epigenomic Landscape of Prokaryotes. PLoS Genet. 2016, 12, e1005854, doi:10.1371/journal.pgen.1005854.
- Wion, D.; Casadesús, J. N6-methyl-adenine: An epigenetic signal for DNA–protein interactions. Nat. Rev. Microbiol. 2006, 4, 183–192, doi:10.1038/nrmicro1350.
- Estibariz, I.; Overmann, A.; Ailloud, F.; Krebes, J.; Josenhans, C.; Suerbaum, S. The core genome m5C methyltransferase JHP1050 (M.Hpy99III) plays an important role in orchestrating gene expression in Helicobacter pylori. Nucleic Acids Res. 2019, 47, 2336–2348, doi:10.1093/nar/gky1307.
- Van der Woude, M.; Braaten, B.; Low, D. Epigenetic phase variation of the pap operon in Escherichia coli. Trends Microbiol. 1996, 4, 5–9, doi:10.1016/0966-842x(96)81498-3.
- Cota, I.; Sánchez-Romero, M.A.; Hernández, S.B.; Pucciarelli, M.G.; García-del Portillo, F.; Casadesús, J. Epigenetic Control of Salmonella enterica O-Antigen Chain Length: A Tradeoff between Virulence and Bacteriophage Resistance. PLoS Genet. 2015, 11, e1005667, doi:10.1371/journal.pgen.1005667.
- Cota, I.; Blanc-Potard, A.B.; Casadesús, J. STM2209-STM2208 (opvAB): A Phase Variation Locus of Salmonella enterica Involved in Control of O-Antigen Chain Length. PLoS ONE 2012, 7, e36863, doi:10.1371/journal.pone.0036863.
- Srikhanta, Y.N.; Maguire, T.L.; Stacey, K.J.; Grimmond, S.M.; Jennings, M.P. The phasevarion: A genetic system controlling coordinated, random switching of expression of multiple genes. Proc. Natl. Acad. Sci. USA 2005, 102, 5547–5551, doi:10.1073/pnas.0501169102.
- De Bolle, X.; Bayliss, C.D.; Field, D.; Van De Ven, T.; Saunders, N.J.; Hood, D.W.; Moxon, E.R. The length of a tetranucleotide repeat tract in Haemophilus influenzae determines the phase variation rate of a gene with homology to type III DNA methyltransferases. Mol. Microbiol. 2000, 35, 211–222, doi:10.1046/j.1365-2958.2000.01701.x.
- De Vries, N.; Duinsbergen, D.; Kuipers, E.J.; Pot, R.G.J.; Wiesenekker, P.; Penn, C.W.; van Vliet, A.H.M.; Vandenbroucke-Grauls, C.M.J.E.; Kusters, J.G. Transcriptional Phase Variation of a Type III Restriction-Modification System in Helicobacter pylori. J. Bacteriol. 2002, 184, 6615–6623, doi:10.1128/jb.184.23.6615-6624.2002.
- Atack, J.M.; Yang, Y.; Seib, K.L.; Zhou, Y.; Jennings, M.P. A survey of Type III restriction-modification systems reveals numerous, novel epigenetic regulators controlling phase-variable regulons; phasevarions. Nucleic Acids Res. 2018, 46, 3532–3542, doi:10.1093/nar/gky192.
- Anjum, A.; Brathwaite, K.J.; Aidley, J.; Connerton, P.L.; Cummings, N.J.; Parkhill, J.; Connerton, I.; Bayliss, C.D. Phase variation of a Type IIG restriction-modification enzyme alters site-specific methylation patterns and gene expression in Campylobacter jejuni strain NCTC11168. Nucleic Acids Res. 2016, 44, 4581–4594, doi:10.1093/nar/gkw019.
- De Ste Croix, M.; Vacca, I.; Kwun, M.J.; Ralph, J.D.; Bentley, S.D.; Haigh, R.; Croucher, N.J.; Oggioni, M.R. Phase-variable methylation and epigenetic regulation by type I restriction–modification systems. FEMS Microbiol. Rev. 2017, 41, S3-S15, doi:10.1093/femsre/fux025.
- Sánchez-Romero, M.A.; Casadesús, J. The bacterial epigenome. Nat. Rev. Microbiol. 2020, 18, 7–20, doi:10.1038/s41579-019-0286-2.