Hepatitis B virus (HBV) is a globally-distributed pathogen and is a major cause of liver disease. HBV (or closely-related animal hepadnaviruses) can integrate into the host genome, but (unlike retroviruses) this integrated form is replication-defective.
- hepatitis B virus
- HBV DNA integration
- hepatocellular carcinoma (HCC)
1. Introduction
Chronic infection with the human hepatitis B virus (HBV) is one of the major drivers of liver disease and is the most common cause of liver cancer worldwide. Approximately one third of the world’s population has been exposed to the virus, and ~257 million people currently live with a chronic HBV infection [1][2][1,2]. Chronic HBV infections are usually life-long (with few exceptions) as the virus has developed several persistence mechanisms to escape immune surveillance, including replication via a highly-stable nuclear episomal template (cccDNA) mimicking a mini-chromosome. Another mode of escaping elimination by the immune systems is the expression of sub-viral particles that dampen antiviral immune responses by mechanisms not yet fully understood.
2. Natural History of Chronic Hepatitis B
Chronic HBV infections are generally asymptomatic for long periods (up to decades) as the virus replicates without triggering an antiviral immune response [3]. Decades later, a sub-optimal immune response is raised against HBV-expressing hepatocytes through yet-unknown mechanisms. The host response is usually inadequate to overcome viral persistence mechanisms, but instead leads to chronic inflammation, liver damage, and hepatic disease.
Surveillance and ongoing care for chronic HBV infection is complicated by psychosocial factors, including limited access to or distrust of the health care system, financial burdens, social stigma, and systemic discrimination [4]. A poor cascade of care results: only ~10% of cases are diagnosed and only ~3% are adequately treated even in some developed countries [5][6][5,6]. Thus, HBV-related disease generally progresses unmonitored and kills ~880,000 people annually through liver cirrhosis and liver cancer [1][2][1,2].
The mechanisms by which HBV causes liver cancer are still not well understood and are under intense investigation. One of the major factors thought to be involved in hepatocarcinogenesis is the integration of the viral DNA genome into cellular chromosomes. This reportedly induces pro-oncogenic pathways through several means (previously reviewed in detail [7][8][9][7,8,9]), including cis-mediated mechanisms (e.g., HBV DNA integrations modulating expression of proximal cellular genes) or trans-mediated mechanisms (e.g., chronic expression of particular viral antigens). This review focuses on how in vitro models have helped in furthering our understanding of how integrated HBV DNA contributes to virus persistence and pathogenesis.
3. HBV Structure
HBV is the prototypical member of the Hepadnaviridae family, which are small enveloped, hepatotropic DNA viruses (~3.2 kbp) replicating via reverse transcription. The double-stranded DNA genome of HBV can take two forms (Figure 1): relaxed circular DNA (rcDNA, generally present in ~90% of virions [10]) or double stranded linear DNA (dslDNA, ~10% of virions). Unlike the fully replication-competent relaxed circular form, the dslDNA-containing viral particles are replication-defective (detailed further below) but can integrate via non-homologous recombination into hepatocyte chromosomes. The HBV genome contains four overlapping open reading frames that encode for seven viral proteins, including: the HBV core antigen (HBcAg, which forms the viral capsid), e antigen (HBeAg, a secreted viral protein encoded by the core/pre-core ORF with reported tolerogenic properties [11]), surface protein (HBsAg, which has three forms large, medium and small—or LHBs, MHBs, and SHBs—that envelop the virion), polymerase (HBV pol, which is essential for viral reverse transcription and replication), and the x protein (HBx, which regulates transcription of viral genes from the cccDNA episome).
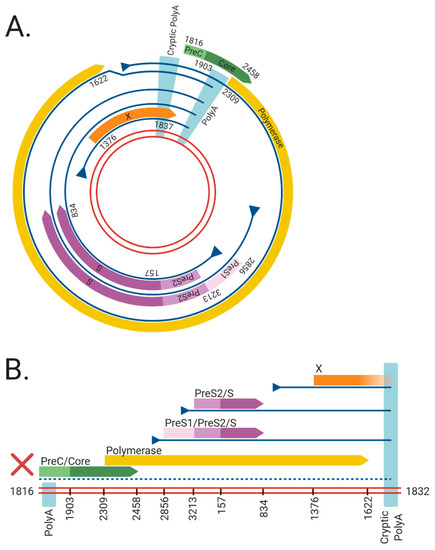
Figure 1. The viral RNAs and open reading frames (ORFs) expressed from the (A) cccDNA and (B) dslDNA forms of hepatitis B virus (HBV). The double red lines represent of the HBV DNA genome, blue lines represent viral mRNAs, triangles indicate transcriptional promotors, and coloured arrows represent viral ORFs. Nucleotide numbering of ORFs are shown as per Genbank Accession #AB241115. The viral mRNAs expressed from cccDNA terminate at a polyadenylation signal (poly A) located in the Core ORF. However, in dslDNA form, the canonical poly A signal is located at the 5′ end and mRNAs instead terminate at a non-canonical cryptic poly A signal. The mRNA coding for the HBV PreC/Core and Polymerase are separated from its promoter in the dslDNA form, leading to loss of their expression (dashed line). Figure generated using BioRender (https://biorender.com/).
4. HBV Replication
HBV replication starts with rcDNA-containing HBV particles attaching to heparan sulphate proteoglycans on the surface of hepatocytes [12][13][14][12,13,14] (Figure 2). This attachment sets up conditions for a high-affinity interaction between the preS-domain of LHBs and sodium taurocholate co-transporting polypeptide (NTCP), which acts as the functional receptor for HBV [15][16][15,16]. Binding is reportedly followed by clathrin-mediated endocytosis of the viral particle [17][18][17,18] and cytoplasmic release of the HBV nucleocapsid, which is subsequently transported to the nucleus and disassembles at the nuclear pore complex [19].

Figure 2. Replication cycle of hepatitis B virus (HBV) including integration into host genome. HBV virions attach to heparan sulphate proteoglycans (HSPG) on the cell surface, allowing for clathrin-mediated endocytosis via sodium taurochlorate co-transporting polypeptide (NTCP). Relaxed circular (rc)DNA genomes (top) are converted into covalently closed circular (ccc)DNA, which serve as the transcriptional template for viral mRNAs (vRNAs) and pregenomic (pg)RNA. Double stranded linear (dsl)DNA (bottom) can also form cccDNA but cannot code for functional rcDNA due to an additional 16nt insertion (asterisk) [20][21][22]. HBV pgRNA is encapsidated by viral capsid proteins and is reverse transcribed by the viral polymerase to produce either rcDNA or dslDNA. The mature nucleocapsids containing viral DNA are then enveloped by HBsAg embedded into host membranes and secreted as virions. As a secondary pathway, dslDNA can integrate into the host genome at double stranded DNA breaks, via non-homologous end joining. Integrated HBV DNA is replication-deficient due to rearrangements of the viral genome that abrogate expression of the viral polymerase and capsid proteins (
The HBV rcDNA genome is released into the nucleus, where it is repaired into the covalently closed circular DNA (cccDNA) form by multiple host DNA repair enzymes [24][25][26][27][28][29][30][31][24,25,26,27,28,29,30,31]. cccDNA acts as the template for all viral transcripts including pregenomic RNA (pgRNA), which is reverse-transcribed in nucleocapsids formed by the viral core antigen. This results in HBV DNA-containing nucleocapsids that are enveloped by host membranes studded with all three forms of HBsAg and then secreted from the cell via multi-vesicular bodies as virions.
5. HBV dslDNA and HBV DNA Integration
While the majority of intra-capsid reverse transcription events result in the formation of rcDNA, a minority of virions contain HBV dslDNA (though this ratio can fluctuate in patients in different stages of infection [10]). The production of dslDNA is dependent on binding and circularisation signals present on the reverse-transcribed strand of HBV DNA [32][33][32,33]. When dslDNA-containing virions infect a cell, HBV genome (instead of being converted into cccDNA) can integrate into cellular genome at the site of cellular double-stranded DNA breaks [34].
The role of integrated HBV DNA is currently unclear: unlike retroviruses, the integrated form of HBV is replication-deficient because of rearrangements that abrogate the expression of capsid, polymerase and functional pgRNA (Figure 1). Firstly, the orientation of the HBV dslDNA separates the HBcAg and pol at its 5′ end from their native promoter at its 3′ end. Moreover, the poly A signal shared by all viral transcripts is located on the 5′ end instead of the 3′ end of the dslDNA, leading to truncated transcripts lacking a canonical poly A. Most viral transcripts expressed from integrated HBV DNA can retain function by terminating using a non-canonical poly A signal retained at its 3′ end [35][36][35,36]. However, pgRNA is rendered non-functional as the premature termination removes structural elements from the 3′ end necessary for initiating pgRNA reverse transcription (namely, the direct repeat 1 and phi sequences [37][38][39][40][37,38,39,40]). Finally, terminal truncations on both ends of the integrated genome are introduced by the error-prone host DNA repair pathways during HBV DNA integration and can disrupt viral ORFs [34][41][42][43][34,41,42,43]. Together, these effects render integrated HBV DNA replication-deficient. No known cell lines with integrated HBV DNA derived from a natural infection produce infectious virus, though they can still express functional HBsAg [44][45][46][47][44,45,46,47].
Various functions have been ascribed to the integrated HBV DNA form (e.g., supporting viral persistence and pathogenesis). In vitro models (Table 1) have enabled detailed understanding of these aspects, including: the molecular mechanisms governing HBV DNA integration; its role as a source of HBsAg in a chronic infection; and how it is involved in HCC initiation and progression. The remainder of this review summarises the knowledge that these model systems have provided the field.
6. In Vitro Models of HBV DNA Integration
In vitro models vary in their ease of use (or reproducibility) in integration studies and inversely how closely they resemble integrations from a true infection in people with HBV. Tumour-derived cell lines from HBV patients contain integrations formed in the native setting but cannot be used to understand underlying mechanisms as they do not generate new integrations. Conversely, nuclear introduction of large numbers of HBV DNA molecules (via over-expressing constructs or transfection) can drive relatively high integration rates, allowing easy study of the integration process. However, this does not reflect a true infection; nuclear import of new HBV virions occurs relatively inefficiently in HBV infection [48][49][50][51][48,49,50,51], so levels of HBV DNA in infected hepatocytes is relatively low. Newly-developed infection models expressing the HBV receptor have solved this to some degree by largely recapitulating the virological aspects accurately. However, the low integration rate is challenging to detect without specialised methods, which can be difficult to reproduce. In essence, all studies need to be interpreted with these limitations in mind.
Table 1. A summary of in vitro model systems for human HBV DNA integration.
Model System | Type | Infectious HBV Produced? | Forms New HBV DNA Integrations? | Refs | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
PLC/PRF/5 | Tumour-derived cell line 1 | No | No | |||||||||||
Hep3B | Tumour-derived cell line 1 | No | No | |||||||||||
HepG2.2.15 | Engineered HBV-producer cell line 2 | Yes | Yes | |||||||||||
HepAD38 | Engineered HBV-producer cell line 2 | Yes | Unknown |
[54] |
||||||||||
Transfection of HBV over-length constructs | HBV transfection 3 | Yes | Unknown | |||||||||||
Transfection of HBV monomeric DNA | HBV transfection 3 | Yes | Unknown | |||||||||||
Transfection of HBV virion DNA | HBV transfection 3 | Yes | Yes |
[62] |
||||||||||
Transfection of in vitro transcribed HBV pgRNA | HBV transfection 3 | Yes | Unknown |
[63] |
||||||||||
Huh7-NTCP | HBV infection 4 | Yes | Yes | |||||||||||
HepG2-NTCP | HBV infection 4 | Yes | Yes | |||||||||||
HepaRG | HBV infection 4 | Yes | Yes | |||||||||||
HepaRG-NTCP | HBV infection 4 | Yes | Yes | |||||||||||
Primary human hepatocytes | HBV infection 4 | Yes | Yes |
1 Tumour-derived cell lines contain replication-deficient integrated HBV DNA acquired during infection. 2 Engineered HBV-producer cell lines contain stable recombinant replication-competent HBV constructs. 3 HBV transfection models involve ectopic introduction of viral DNA or RNA constructs. 4 HBV infection models involve bona fide viral infection of cells expressing the viral receptor NTCP.