Acute promyelocytic leukemia (APL) represents a paradigm of precision medicine.
- acute promyelocytic leukemia
- children
1. Epidemiology and Biology of Acute Promyelocytic Leukemia in Childhood
Acute promyelocytic leukemia (APL) is a unique subtype of acute myeloid leukemia (AML) accounting for 5–10% of all pediatric AML cases [1].
A higher incidence has been described in Italy, Spain, China, and Latin America, with the highest proportions of APL (up to 50% of all AML cases) reported in Nicaragua and Argentina [2,3,4]. The incidence becomes higher with age with a peak in the fourth decade, while APL is particularly rare in infants (median age of pediatric cases is 9–12 years) [5,6]. Data from a population-based study of SEER (Surveillance, Epidemiology, and End Results Program) on demographics and epidemiology of 1397 patients with APL diagnosed between 1975 and 2008 in the United States showed an incidence of 0.06 per 100,000 persons/year in the pediatric population (aged ≤ 20 years) [7]. Moreover, APL occurs equally in both males and females, and obesity has been recognized as a possible risk factor [8].
A higher incidence has been described in Italy, Spain, China, and Latin America, with the highest proportions of APL (up to 50% of all AML cases) reported in Nicaragua and Argentina [2][3][4]. The incidence becomes higher with age with a peak in the fourth decade, while APL is particularly rare in infants (median age of pediatric cases is 9–12 years) [5][6]. Data from a population-based study of SEER (Surveillance, Epidemiology, and End Results Program) on demographics and epidemiology of 1397 patients with APL diagnosed between 1975 and 2008 in the United States showed an incidence of 0.06 per 100,000 persons/year in the pediatric population (aged ≤ 20 years) [7]. Moreover, APL occurs equally in both males and females, and obesity has been recognized as a possible risk factor [8].
Considered to be a rare disease, childhood APL has been traditionally treated borrowing adult protocols and, only in the last two decades, dedicated pediatric protocols have been designed.
After the first description in 1957 [9], in 1977 Rowley and colleagues identified the typical balanced chromosomal translocation t(15;17)(q22;q21). This represents a pathogenetic hallmark of APL and is detected in 95–98% of cases [10]. This translocation, joining the promyelocytic leukemia (PML) gene on chromosome 15 to the retinoic acid receptor alpha (RARA) gene on chromosome 17, leads to an oncogenic fusion gene (PML-RARA) [11]. In particular, the breakpoint of the RARA gene is located in its second intron, while three different breakpoints of the PML gene (intron 6, exon 6, and intron 3) identify the most common breakpoint cluster regions (bcr-1, -2, and -3, respectively), corresponding to different PML-RARA isoforms [12]. While bcr-1 (also known as “long” or “L” isoform) and bcr-3 (also known as “short” or “S” isoform) account for the majority of APL cases (90–95%), bcr-2 represents a more rare variant (also called “V”) [13] and no particular differences (apart from those related to ethnicity) are known as to the different frequency of these isoforms between children and adults [14,15].
After the first description in 1957 [9], in 1977 Rowley and colleagues identified the typical balanced chromosomal translocation t(15;17)(q22;q21). This represents a pathogenetic hallmark of APL and is detected in 95–98% of cases [10]. This translocation, joining the promyelocytic leukemia (PML) gene on chromosome 15 to the retinoic acid receptor alpha (RARA) gene on chromosome 17, leads to an oncogenic fusion gene (PML-RARA) [11]. In particular, the breakpoint of the RARA gene is located in its second intron, while three different breakpoints of the PML gene (intron 6, exon 6, and intron 3) identify the most common breakpoint cluster regions (bcr-1, -2, and -3, respectively), corresponding to different PML-RARA isoforms [12]. While bcr-1 (also known as “long” or “L” isoform) and bcr-3 (also known as “short” or “S” isoform) account for the majority of APL cases (90–95%), bcr-2 represents a more rare variant (also called “V”) [13] and no particular differences (apart from those related to ethnicity) are known as to the different frequency of these isoforms between children and adults [14][15].
In normal conditions, PML interacts with p53 and it is critical for cellular senescence and tumor suppression activities, while RARA interacts as a heterodimer with RXR and regulates transcription factors important for myeloid differentiation [16]. In APL, the PML-RARA fusion oncoprotein is responsible for the myeloid differentiation block at the promyelocyte stage [17]. The combination of all-trans retinoid acid (ATRA) and arsenic trioxide (ATO) allows the proteasomal degradation of the oncoprotein via its ubiquitination and finally reactivates the transcription of RARA target genes [18,19,20] (
In normal conditions, PML interacts with p53 and it is critical for cellular senescence and tumor suppression activities, while RARA interacts as a heterodimer with RXR and regulates transcription factors important for myeloid differentiation [16]. In APL, the PML-RARA fusion oncoprotein is responsible for the myeloid differentiation block at the promyelocyte stage [17]. The combination of all-trans retinoid acid (ATRA) and arsenic trioxide (ATO) allows the proteasomal degradation of the oncoprotein via its ubiquitination and finally reactivates the transcription of RARA target genes [18][19][20] (
). Other mechanisms of action include the recruitment of caspases, the production of reactive oxygen species, and the reformation of PML-nuclear bodies, whose architecture is disrupted by PML-RARA fusion protein with critical consequences for normal cellular functions, such as self-renewal [21].
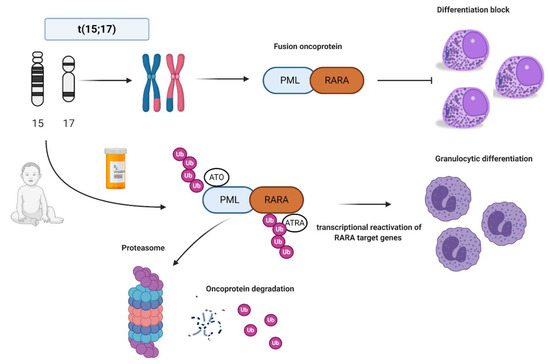
Acute promyelocytic leukemia (APL) biology and all-trans retinoic acid/arsenic trioxide (ATRA/ATO) synergistic treatment. APL is the result of the balanced translocation between promyelocytic leukemia (PML) gene on chromosome 15 and retinoic acid receptor alpha (RARA) on chromosome 17 leading to the fusion oncoprotein PML-RARA and myeloid differentiation block at promyelocyte stage. The combination of ATRA and ATO leads to ubiquitination and, ultimately, to proteasome degradation of the fusion protein, reactivation of the transcription of RARA target genes, and finally granulocytic differentiation. Images were generated using BioRender.
2. Diagnostics, Clinical Features, and Risk Assessment
As aforementioned, the classical hallmark of APL is the cytogenetic translocation t(15;17), present in 95–98% of cases, and detectable by the specific fluorescence in situ hybridization (FISH) probes, and more rapid molecular biology techniques such as reverse transcriptase polymerase chain reaction (RT-PCR). However, a small percentage of cases presents different RARA fusion partners, such as nucleophosmin (NPM1, 5q35), signal transducer, and activator of transcription (STAT5B, 17q21), factor interacting with PAPOLA and CPSF1 (FIP1L1, 4q12), promyelocytic leukemia zinc finger (PLZF, now known as ZBTB16, 11q23) and nuclear matrix-mitotic apparatus protein 1 (NUMA1, 11q13) [22,23,24,25,26]. These rare cases of “APL variant” are characterized by the same clinical picture of classic APL but different sensitivity to ATRA/ATO combination [27,28] (
As aforementioned, the classical hallmark of APL is the cytogenetic translocation t(15;17), present in 95–98% of cases, and detectable by the specific fluorescence in situ hybridization (FISH) probes, and more rapid molecular biology techniques such as reverse transcriptase polymerase chain reaction (RT-PCR). However, a small percentage of cases presents different RARA fusion partners, such as nucleophosmin (NPM1, 5q35), signal transducer, and activator of transcription (STAT5B, 17q21), factor interacting with PAPOLA and CPSF1 (FIP1L1, 4q12), promyelocytic leukemia zinc finger (PLZF, now known as ZBTB16, 11q23) and nuclear matrix-mitotic apparatus protein 1 (NUMA1, 11q13) [22][23][24][25][26]. These rare cases of “APL variant” are characterized by the same clinical picture of classic APL but different sensitivity to ATRA/ATO combination [27][28] (
).
ATRA/ATO sensitivity data of the various RARA rearrangements mentioned in the text.
| RARA Rearrangements | Cytogenetic Translocations | ATRA Sensitivity | ATO Sensitivity |
|---|
| PML-RARA | t(15;17)(q22;q21) | S | S |
| FIP1L1-RARA | t(4;17)(q12;q21) | I | NA |
| NPM1-RARA | t(5;17)(q35;q21) | S | NA |
| NuMA-RARA | t(11;17)(q13;q21) | S | NA |
| STAT5b-RARA | t(17;17)(q21;q21) | R | R |
| ZBTB16-RARA | t(11;17)(q23;q21) | R | R |
R, resistant; I, intermediate sensitivity; S, sensitive; NA, data not available.
The presence of Auer rods and hypergranular promyelocyte blast cells is typical of APL. Frequently, the bone marrow classical morphology reveals promyelocytes with abundant Auer rods in multiple clusters (the so-called “faggot cells”). In children, the higher incidence of the microgranular variant (M3v) is controversial. Indeed, in pediatric APL, a higher incidence of the M3v has been reported (30% of pediatric APL vs. 15% in the adult population [29]), but these data have not been confirmed by other studies [29,30,31]. Although the confirmation of the diagnosis is typically based on PCR-based techniques, the immunophenotype study of classic APL is characteristic with the absence or low expression of HLA-DR, CD34, CD11a, and CD11b, bright expression of CD33, and heterogeneous expression of CD13 and CD117 in most cases. MPO is always positive with a high side scatter [29]. Conversely, M3v is frequently associated with the expression of CD2 and CD34 [29,32].
The presence of Auer rods and hypergranular promyelocyte blast cells is typical of APL. Frequently, the bone marrow classical morphology reveals promyelocytes with abundant Auer rods in multiple clusters (the so-called “faggot cells”). In children, the higher incidence of the microgranular variant (M3v) is controversial. Indeed, in pediatric APL, a higher incidence of the M3v has been reported (30% of pediatric APL vs. 15% in the adult population [29]), but these data have not been confirmed by other studies [29][30][31]. Although the confirmation of the diagnosis is typically based on PCR-based techniques, the immunophenotype study of classic APL is characteristic with the absence or low expression of HLA-DR, CD34, CD11a, and CD11b, bright expression of CD33, and heterogeneous expression of CD13 and CD117 in most cases. MPO is always positive with a high side scatter [29]. Conversely, M3v is frequently associated with the expression of CD2 and CD34 [29][32].
Despite its inclusion in the last European LeukemiaNet (ELN) 2017 recommendations under the umbrella of favorable/low-risk AML subtype together with the core-binding factor (CBF) leukemias, APL represents a unique disease with peculiarities in terms of clinical features and risk assessment [33].
The classical onset with a life-threatening coagulopathy characterized by severe and extended hemorrhages makes APL one of the most challenging hematological emergencies, which can lead the patient to death unless an adequate treatment is started [34]. APL blasts express fibrinolytic proteins, procoagulant factors, and proteolytic enzymes and have an increased ability to adhere to the vascular endothelium and secrete inflammatory cytokines, which ultimately stimulate the expression of prothrombotic proteins by leukocytes and endothelial cells [35]. This thrombo-hemorrhagic disturbance creates a clinical picture resembling disseminated intravascular coagulation, which can have different degrees of severity, ranging from laboratory alterations to life-threatening situations. The problem of early deaths (ED), defined as deaths occurring in the first 30 days after diagnosis, is, nowadays, one of the hindrances towards the cure of APL patients [36]. ED have been described to occur in approximately 10% of APL patients treated with ATRA plus chemotherapy in large cooperative trials; however, registry studies suggest a possible discrepancy with the “real world” situation, in which an ED rate up to 29% has been reported [37]. The leading causes of death are intracranial (as high as 70% of cases) followed by intrapulmonary hemorrhage [38,39]. In children, serious bleeding episodes have been reported in up to 15% of cases, and up to 10% of fatal outcomes in some series [40,41,42]. ED has been associated with a high white blood cells (WBC) count. In fact, WBC count at diagnosis is the strongest predictor of outcome and relapse in APL, so that patients are stratified according to the Sanz-risk criteria as standard risk (SR) and high risk (HR), based on the presence of less or more than 10,000/μL WBC, respectively [43].
The classical onset with a life-threatening coagulopathy characterized by severe and extended hemorrhages makes APL one of the most challenging hematological emergencies, which can lead the patient to death unless an adequate treatment is started [34]. APL blasts express fibrinolytic proteins, procoagulant factors, and proteolytic enzymes and have an increased ability to adhere to the vascular endothelium and secrete inflammatory cytokines, which ultimately stimulate the expression of prothrombotic proteins by leukocytes and endothelial cells [35]. This thrombo-hemorrhagic disturbance creates a clinical picture resembling disseminated intravascular coagulation, which can have different degrees of severity, ranging from laboratory alterations to life-threatening situations. The problem of early deaths (ED), defined as deaths occurring in the first 30 days after diagnosis, is, nowadays, one of the hindrances towards the cure of APL patients [36]. ED have been described to occur in approximately 10% of APL patients treated with ATRA plus chemotherapy in large cooperative trials; however, registry studies suggest a possible discrepancy with the “real world” situation, in which an ED rate up to 29% has been reported [37]. The leading causes of death are intracranial (as high as 70% of cases) followed by intrapulmonary hemorrhage [38][39]. In children, serious bleeding episodes have been reported in up to 15% of cases, and up to 10% of fatal outcomes in some series [40][41][42]. ED has been associated with a high white blood cells (WBC) count. In fact, WBC count at diagnosis is the strongest predictor of outcome and relapse in APL, so that patients are stratified according to the Sanz-risk criteria as standard risk (SR) and high risk (HR), based on the presence of less or more than 10,000/μL WBC, respectively [43].
3. Pitfalls and Hurdles of Childhood APL: Relapses and Long-Term
Sequelae
With the use of ATRA and ATO combination, APL relapses have become a rare entity, also in children [82]. Molecular studies on relapsed APL have been typically conducted using adult patient samples and helped to identify the molecular landscape of ATRA-resistant clones retaining self-renewal activities and the contribution of other genes, such as WT1, FLT3, and PML mutations [83,84]. No specific trials have systematically addressed the appropriate therapy and management of relapses and future prospective trials are probably not feasible, especially in children, because of the rarity of the event. However, sporadic case reports showed that ATO may be used for achieving a second CR in pediatric cases, underlying that ATO resistance is almost absent, with the exception of the aforementioned cases carrying PML mutations, with A216V as the predominant lesion [85]. Moreover, GO, an anti-CD33 humanized monoclonal antibody linked to the cytotoxic agent calicheamicin, has also been used with successful results and is approved for children under two years old, at doses lower than 6 mg/m
With the use of ATRA and ATO combination, APL relapses have become a rare entity, also in children [44]. Molecular studies on relapsed APL have been typically conducted using adult patient samples and helped to identify the molecular landscape of ATRA-resistant clones retaining self-renewal activities and the contribution of other genes, such as WT1, FLT3, and PML mutations [45][46]. No specific trials have systematically addressed the appropriate therapy and management of relapses and future prospective trials are probably not feasible, especially in children, because of the rarity of the event. However, sporadic case reports showed that ATO may be used for achieving a second CR in pediatric cases, underlying that ATO resistance is almost absent, with the exception of the aforementioned cases carrying PML mutations, with A216V as the predominant lesion [47]. Moreover, GO, an anti-CD33 humanized monoclonal antibody linked to the cytotoxic agent calicheamicin, has also been used with successful results and is approved for children under two years old, at doses lower than 6 mg/m
2 [86]. Therefore, patients achieving molecular remission may proceed to autologous stem cell transplant, while patients unable to clear the molecular transcript should receive an allogeneic stem cell transplant [82]. Finally, tamibarotene, a synthetic retinoid analog, can overcome ATRA resistance thanks to higher PML-RARA binding affinity, and appears to be promising in the relapsed/refractory setting [87].
[48]. Therefore, patients achieving molecular remission may proceed to autologous stem cell transplant, while patients unable to clear the molecular transcript should receive an allogeneic stem cell transplant [44]. Finally, tamibarotene, a synthetic retinoid analog, can overcome ATRA resistance thanks to higher PML-RARA binding affinity, and appears to be promising in the relapsed/refractory setting [49].
Another important problem in the field of childhood APL is long-term
sequelae. If the occurrence of cardiac toxicity seems to be avoided with the newer anthracycline-free regimens, prolonged arsenic exposure may lead to skin lesion (hyperpigmentation, keratosis, and squamous cell carcinoma), hypertension, diabetes mellitus, neurologic effects, and urinary tract cancers (mainly involving the bladder) [88]. The results of arsenic elimination pharmacokinetics of the study by Zhou et al. and Li et al. seem to be promising, at least demonstrating that arsenic does not last more than 24 months above the safety threshold set by the regulatory agencies [69,71]. Moreover, the post-remission treatment is now based on shorter ATO exposure (less than eight months as compared with the three year post-remission strategy of the first Chinese study). Finally, in children, arsenic might cause an alteration of growth and development, although available data are immature for evaluating long-term toxicities and we need to wait for the results of the ongoing studies. However, the recent data of the oral formulation, showing a better toxicity profile and simpler administration modality as compared with the intravenous formulation, seem to be particularly attractive for the younger population [77].
. If the occurrence of cardiac toxicity seems to be avoided with the newer anthracycline-free regimens, prolonged arsenic exposure may lead to skin lesion (hyperpigmentation, keratosis, and squamous cell carcinoma), hypertension, diabetes mellitus, neurologic effects, and urinary tract cancers (mainly involving the bladder) [50]. The results of arsenic elimination pharmacokinetics of the study by Zhou et al. and Li et al. seem to be promising, at least demonstrating that arsenic does not last more than 24 months above the safety threshold set by the regulatory agencies [51][52]. Moreover, the post-remission treatment is now based on shorter ATO exposure (less than eight months as compared with the three year post-remission strategy of the first Chinese study). Finally, in children, arsenic might cause an alteration of growth and development, although available data are immature for evaluating long-term toxicities and we need to wait for the results of the ongoing studies. However, the recent data of the oral formulation, showing a better toxicity profile and simpler administration modality as compared with the intravenous formulation, seem to be particularly attractive for the younger population [53].
References
- Testi, A.M.; Pession, A.; Diverio, D.; Grimwade, D.; Gibson, B.; de Azevedo, A.C.; Moran, L.; Leverger, G.; Elitzur, S.; Hasle, H.; et al. Risk-adapted treatment of acute promyelocytic leukemia: Results from the International Consortium for Childhood APL. Blood 2018, 132, 405–412.
- Maule, M.; Dama, E.; Mosso, M.L.; Magnani, C.; Pastore, G.; Merletti, F. High incidence of acute promyelocytic leukemia in children in northwest Italy, 1980–2003: A report from the Childhood Cancer Registry of Piedmont. Leukemia 2008, 22, 439–441.
- Corea, A.M.; Espinoza, C.P.; Rajnoldi, A.C.; Conter, V.; Lietti, G.; Masera, G.; Sessa, C.; Cavalli, F.; Biondi, A.; Rovelli, A. Childhood acute promyelocytic leukemia in Nicaragua. Ann. Oncol. 1993, 4, 892–894.
- Gómez, S.M.; Schuttenberg, V.; Armendariz, H.; Alba, L.; Martinez, M.; Fynn, A.; Ferrère, E.; Delgado-Caffé, A. Childhood acute leukemia: A single institution experience in La Plata, Argentina. Med. Pediatr. Oncol. 2001, 36, 383–385.
- Creutzig, U.; Zimmermann, M.; Reinhardt, D.; Rasche, M.; von Neuhoff, C.; Alpermann, T.; Dworzak, M.; Perglerová, K.; Zemanova, Z.; Tchinda, J.; et al. Changes in cytogenetics and molecular genetics in acute myeloid leukemia from childhood to adult age groups. Cancer 2016, 122, 3821–3830.
- Testi, A.M.; Coco, F.L.; D’Angiò, M.; Locatelli, F.; Pession, A. Acute promyelocytic leukemia (APL): Comparison between children and adults. Mediterr. J. Hematol. Infect. Dis. 2014, 6, e2014032.
- Chen, Y.; Kantarjian, H.; Wang, H.; Cortes, J.; Ravandi, F. Acute promyelocytic leukemia: A population-based study on incidence and survival in the United States, 1975–2008. Cancer 2012, 118, 5811–5818.
- Mazzarella, L.; Botteri, E.; Matthews, A.; Gatti, E.; Di Salvatore, D.; Bagnardi, V.; Breccia, M.; Montesinos, P.; Bernal, T.; Gil, C.; et al. Obesity is a risk factor for acute promyelocytic leukemia: Evidence from population and cross-sectional studies and correlation with FLT3 mutations and polyunsaturated fatty acid metabolism. Haematologica 2020, 105, 1559–1566.
- Hillestad, L.K. Acute promyelocytic leukemia. Acta Med. Scand. 1957, 159, 189–194.
- Rowley, J.; Golomb, H.; Dougherty, C. 15/17 translocation, a consistent chromosomal change in acute promyelocytic leukaemia. Lancet 1977, 309, 549–550.
- Lo-Coco, F.; Hasan, S.K. Understanding the molecular pathogenesis of acute promyelocytic leukemia. Best Pr. Res. Clin. Haematol. 2014, 27, 3–9.
- Pandolfi, P.; Alcalay, M.; Fagioli, M.; Zangrilli, D.; Mencarelli, A.; Diverio, D.; Biondi, A.; Coco, F.L.; Rambaldi, A.; Grignani, F. Genomic variability and alternative splicing generate multiple PML/RAR alpha transcripts that encode aberrant PML proteins and PML/RAR alpha isoforms in acute promyelocytic leukaemia. EMBO J. 1992, 11, 1397–1407.
- Liquori, A.; Ibañez, M.; Simarro, C.S.; Sanz, M.A.; Barragán, E.; Cervera, J. Acute promyelocytic leukemia: A constellation of molecular events around a single PML-RARA fusion gene. Cancers 2020, 12, 624.
- Conneely, S.E.; Stevens, A.M. Advances in pediatric acute promyelocytic leukemia. Children 2020, 7, 11.
- Zhao, J.; Liang, J.-W.; Xue, H.-L.; Shen, S.-H.; Chen, J.; Tang, Y.-J.; Yu, L.-S.; Liang, H.-H.; Gu, L.-J.; Tang, J.; et al. The genetics and clinical characteristics of children morphologically diagnosed as acute promyelocytic leukemia. Leukemia 2018, 33, 1387–1399.
- Guo, A.; Salomoni, P.; Luo, J.; Shih, A.; Zhong, S.; Gu, W.; Pandolfi, P.P. The function of PML in p53-dependent apoptosis. Nat. Cell Biol. 2000, 2, 730–736.
- Dos Santos, G.A.; Kats, L.; Pandolfi, P.P. Synergy against PML-RARa: Targeting transcription, proteolysis, differentiation, and self-renewal in acute promyelocytic leukemia. J. Exp. Med. 2013, 210, 2793–2802.
- Lallemand-Breitenbach, V.; Jeanne, M.; Benhenda, S.; Nasr, R.; Lei, M.; Peres, L.; Zhou, J.; Raught, B.; de Thé, H. Arsenic degrades PML or PML–RARα through a SUMO-triggered RNF4/ubiquitin-mediated pathway. Nat. Cell Biol. 2008, 10, 547–555.
- Nasr, R.; Guillemin, M.-C.; Ferhi, O.; Soilihi, H.; Peres, L.; Berthier, C.; Rousselot, P.; Robledo-Sarmiento, M.; Lallemand-Breitenbach, V.; Gourmel, B.; et al. Eradication of acute promyelocytic leukemia-initiating cells through PML-RARA degradation. Nat. Med. 2008, 14, 1333–1342.
- Kwok, C.; Zeisig, B.B.; Dong, S.; So, C.W.E. Forced homo-oligomerization of RARα leads to transformation of primary hematopoietic cells. Cancer Cell 2006, 9, 95–108.
- Voisset, E.; Moravcsik, E.; Stratford, E.W.; Jaye, A.; Palgrave, C.J.; Hills, R.K.; Salomoni, P.; Kogan, S.C.; Solomon, E.; Grimwade, D. Pml nuclear body disruption cooperates in APL pathogenesis and impairs DNA damage repair pathways in mice. Blood 2018, 131, 636–648.
- Chen, Z.; Guidez, F.; Rousselot, P.; Agadir, A.; Chen, S.J.; Wang, Z.Y.; Degos, L.; Zelent, A.; Waxman, S.; Chomienne, C. PLZF-RAR alpha fusion proteins generated from the variant t(11;17)(q23;q21) translocation in acute promyelocytic leukemia inhibit ligand-dependent transactivation of wild-type retinoic acid receptors. Proc. Natl. Acad. Sci. USA 1994, 91, 1178–1182.
- Corey, S.J.; Locker, J.; Oliveri, D.R.; Shekhter-Levin, S.; Redner, R.L.; Penchansky, L.; Gollin, S.M. A non-classical translocation involving 17q12 (retinoic acid receptor alpha) in acute promyelocytic leukemia (APML) with atypical features. Leukemia 1994, 8, 1350–1353.
- De Braekeleer, E.; Douet-Guilbert, N.; De Braekeleer, M. RARA fusion genes in acute promyelocytic leukemia: A review. Expert Rev. Hematol. 2014, 7, 347–357.
- Wells, R.A.; Catzavelos, C.; Kamel-Reid, S. Fusion of retinoic acid receptor α to NuMA, the nuclear mitotic apparatus protein, by a variant translocation in acute promyelocytic leukaemia. Nat. Genet. 1997, 17, 109–113.
- Geoffroy, M.-C.; de Thé, H. Classic and variants APLs, as viewed from a therapy response. Cancers 2020, 12, 967.
- Ciangola, G.; Gurnari, C.; Paterno, G.; Mirabile, M.; Angelini, M.; Lavorgna, S.; Ottone, T.; Travaglini, S.; Cicconi, L.; Lo-Coco, F. STAT5b-RARa-positive acute myeloid leukemia: Diagnostic and therapeutic challenges of a rare AML subtype. Leuk. Res. 2019, 78, 21–23.
- Sanz, M.A.; Fenaux, P.; Tallman, M.S.; Estey, E.H.; Löwenberg, B.; Naoe, T.; Lengfelder, E.; Döhner, H.; Burnett, A.K.; Chen, S.-J.; et al. Management of acute promyelocytic leukemia: Updated recommendations from an expert panel of the European LeukemiaNet. Blood 2019, 133, 1630–1643.
- Guglielmi, C.; Martelli, M.P.; Diverio, D.; Fenu, S.; Vegna, M.L.; Cantù-Rajnoldi, A.; Biondi, A.; Cocito, M.G.; Del Vecchio, L.; Tabilio, A.; et al. Immunophenotype of adult and childhood acute promyelocytic leukaemia: Correlation with morphology, type of PML gene breakpoint and clinical outcome. A cooperative Italian study on 196 cases. Br. J. Haematol. 1998, 102, 1035–1041.
- Testi, A.M.; Biondi, A.; Albano, F.; Moleti, M.L.; Giona, F.; Vignetti, M.; Menna, G.; Locatelli, F.; Pession, A.; Barisone, E.; et al. GIMEMA-AIEOPAIDA protocol for the treatment of newly diagnosed acute promyelocytic leukemia (APL) in children. Blood 2005, 106, 447–453.
- Bally, C.; Fadlallah, J.; Leverger, G.; Bertrand, Y.; Robert, A.; Baruchel, A.; Guerci, A.; Récher, C.; Raffoux, E.; Thomas, X.; et al. Outcome of acute promyelocytic leukemia (APL) in children and adolescents: An analysis in two consecutive trials of the European APL Group. J. Clin. Oncol. 2012, 30, 1641–1646.
- Swerdlow, S.H.; Campo, E.; Harris, N.L.; Jaffe, E.S.; Pileri, S.A.; Stein, H.; Thiele, J. (Eds.) WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues, revised 4th ed.; IARC: Lyon, France, 2017; Volume 2.
- Döhner, H.; Estey, E.; Grimwade, D.; Amadori, S.; Appelbaum, F.R.; Büchner, T.; Dombret, H.; Ebert, B.L.; Fenaux, P.; Larson, R.A.; et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood 2017, 129, 424–447.
- Sanz, M.A.; Montesinos, P. Advances in the management of coagulopathy in acute promyelocytic leukemia. Thromb. Res. 2020, 191, S63–S67.
- Falanga, A.; Russo, L.; Tartari, C.J. Pathogenesis and treatment of thrombohemorrhagic diathesis in acute promyelocytic leukemia. Mediterr. J. Hematol. Infect. Dis. 2011, 3, e2011068.
- Gurnari, C.; Breccia, M.; Di Giuliano, F.; Scalzulli, E.; Divona, M.; Piciocchi, A.; Cicconi, L.; De Bellis, E.; Venditti, A.; Del Principe, M.I.; et al. Early intracranial haemorrhages in acute promyelocytic leukaemia: Analysis of neuroradiological and clinico-biological parameters. Br. J. Haematol. 2020. online version. [Google Scholar] [CrossRef]
- Breccia, M.; Lo-Coco, F. Thrombo-hemorrhagic deaths in acute promyelocytic leukemia. Thromb. Res. 2014, 133, S112–S116.
- Naymagon, L.; Moshier, E.; Tremblay, D.; Mascarenhas, J. Predictors of early hemorrhage in acute promyelocytic leukemia. Leuk. Lymphoma 2019, 60, 2394–2403.
- Martinez-Cuadron, D.; Sobas, M.; Vellenga, E.; Fernandez, I.; Sossa, C.; de Lisa, E.; Bernal, T.; Serrano, J.; Salamero, O.; Brunet, S.; et al. PF255 incidence and risk factors for early death among 2421 APL patients: The pethema registry experience. HemaSphere 2019, 3, 78–79.
- Rajpurkar, M.; Alonzo, T.A.; Wang, Y.-C.; Gerbing, R.B.; Gamis, A.S.; Feusner, J.H.; Gregory, J.; Kutny, M.A. Risk markers for significant bleeding and thrombosis in pediatric acute promyelocytic leukemia. Report from the Children’s Oncology Group study AAML0631. J. Pediatr. Hematol. 2019, 41, 51–55.
- De Azevedo, A.C.; Matsuda, E.; Cervellini, J.Y.; Prandi, L.R.; Omae, C.; Jotta, P.Y.; Pereira, R.M.; Brandalise, S.R. Early mortality in children and adolescents with acute promyelocytic leukemia: Experience of the Boldrini Children’s Center. J. Pediatr. Hematol. Oncol. 2020, 42, e641–e646.
- Zhang, Y.; Wang, L.; Zhang, R.; Qi, P.; Xie, J.; Shi, H.; Lin, W.; Wu, Y.; Yu, J.; Fan, J.; et al. Long-term follow-up of children with acute promyelocytic leukemia treated with Beijing Children’s Hospital APL 2005 protocol (BCH-APL 2005). Pediatr. Hematol. Oncol. 2019, 36, 399–409.
- Sanz, M.A.; Lo Coco, F.; Martín, G.; Avvisati, G.; Rayón, C.; Barbui, T.; Díaz-Mediavilla, J.; Fioritoni, G.; González, J.D.; Liso, V.; et al. Definition of relapse risk and role of nonanthracycline drugs for consolidation in patients with acute promyelocytic leukemia: A joint study of the PETHEMA and GIMEMA cooperative groups. Blood 2000, 96, 1247–1253.
- Abla, O.; Kutny, M.A.; Testi, A.M.; Feusner, J.H.; Creutzig, U.; Gregory, J.; Gibson, B.; Leverger, G.; Ribeiro, R.C.; Smith, O.; et al. Management of relapsed and refractory childhood acute promyelocytic leukaemia: Recommendations from an international expert panel. Br. J. Haematol. 2016, 175, 588–601.
- Iaccarino, L.; Ottone, T.; Alfonso, V.; Cicconi, L.; Divona, M.; Lavorgna, S.; Travaglini, S.; Ferrantini, A.; Falconi, G.; Baer, C.; et al. Mutational landscape of patients with acute promyelocytic leukemia at diagnosis and relapse. Am. J. Hematol. 2019, 94, 1091–1097.
- Lehmann-Che, J.; Bally, C.; Letouzé, E.; Berthier, C.; Yuan, H.; Jollivet, F.; Ades, L.; Cassinat, B.; Hirsch, P.; Pigneux, A.; et al. Dual origin of relapses in retinoic-acid resistant acute promyelocytic leukemia. Nat. Commun. 2018, 9, 1–8.
- Lu, J.; Xiao-Jun, H.; Bao, L.; Jiang, H.; Zhu, H.; Jiang, B. Treatment outcomes in relapsed acute promyelocytic leukemia patients initially treated with all-trans retinoic acid and arsenic compound-based combined therapies. Oncol. Lett. 2014, 7, 177–182.
- Wang, E.S.; Aplenc, R.; Chirnomas, D.; Dugan, M.; Fazal, S.; Iyer, S.; Lin, T.L.; Nand, S.; Pierce, K.J.; Shami, P.J.; et al. Safety of gemtuzumab ozogamicin as monotherapy or combination therapy in an expanded-access protocol for patients with relapsed or refractory acute myeloid leukemia. Leuk. Lymphoma 2020, 61, 1965–1973.
- Sanford, D.; Lo-Coco, F.; Sanz, M.A.; Di Bona, E.; Coutre, S.; Altman, J.K.; Wetzler, M.; Allen, S.L.; Ravandi, F.; Kantarjian, H.; et al. Tamibarotene in patients with acute promyelocytic leukaemia relapsing after treatment with all-transretinoic acid and arsenic trioxide. Br. J. Haematol. 2015, 171, 471–477.
- Gurnari, C.; De Bellis, E.; Divona, M.; Ottone, T.; Lavorgna, S.; Voso, M.T. When poisons cure: The case of arsenic in acute promyelocytic leukemia. Chemotherapy 2019, 64, 238–247.
- Zhou, J.; Zhang, Y.; Li, J.; Li, X.; Hou, J.; Zhao, Y.; Liu, X.; Han, X.; Hu, L.; Wang, S.; et al. Single-agent arsenic trioxide in the treatment of children with newly diagnosed acute promyelocytic leukemia. Blood 2010, 115, 1697–1702.
- Zhang, L.; Zou, Y.; Chen, Y.; Guo, Y.; Yang, W.; Chen, X.; Wang, S.; Liu, X.; Ruan, M.; Zhang, J.; et al. Role of cytarabine in paediatric acute promyelocytic leukemia treated with the combination of all-trans retinoic acid and arsenic trioxide: A randomized controlled trial. BMC Cancer 2018, 18, 374.
- Yang, M.-H.; Wan, W.-Q.; Luo, J.-S.; Zheng, M.-C.; Huang, K.; Yang, L.-H.; Mai, H.-R.; Li, J.; Chen, H.-Q.; Sun, X.-F.; et al. Multicenter randomized trial of arsenic trioxide and Realgar-Indigo naturalis formula in pediatric patients with acute promyelocytic leukemia: Interim results of the SCCLG-APL clinical study. Am. J. Hematol. 2018, 93, 1467–1473.