Pancreatic ductal adenocarcinoma (PDAC), is characterized by an overall poor prognosis and a five-year survival that is less than 10% due to late diagnosis, aggressive disease progression, and resistance to conventional chemotherapy. A large proportion of a pancreatic tumor is made up of stromal fibroblasts, the pancreatic stellate cells (PSCs, also known as cancer-associated fibroblasts – CAFs). Increasing evidence indicates that PSCs contribute to the overall tumor progression, drug resistance, and metabolic rewiring in PDAC.
- pancreatic cancer
- pancreatic stellate cell
- metabolic reprogramming
- gemcitabine chemoresistance
1. Introduction
Pancreatic ductal adenocarcinoma (PDAC), commonly referred to as pancreatic cancer (PC), comprises more than 85% of all pancreatic tumors. PDAC is a highly malignant tumor with a notoriously dismal prognosis[1] and ranks among the leading causes of cancer-related deaths in the Western world[2]. The overall five-year survival rate of merely 7% for all stages of the disease is among the lowest of all solid tumor types[3]. Despite certain recent treatment advances such as neoadjuvant treatment followed by surgery and combination chemotherapies using FOLinic acid, 5-Fluorouracil, IRINotecan, and Oxaliplatin (FOLFIRINOX) and/or gemcitabine, the overall prognosis of PDAC patients remains poor[4][5][6].
The dismal clinical outcomes have been linked to the presence of both intrinsic (de novo) and acquired resistance to the existing therapeutics, especially against gemcitabine. Since 1997, gemcitabine has been the gold standard of care for all stages of PDAC[7][8]. One of the main challenges of PDAC treatment lies in the fact that cytotoxic drugs may achieve good results in preclinical test models, however, they generally fail to do so when tested clinically. The main culprit is the presence of an exceedingly prominent stroma in PDAC, which acts as a mechanical barrier to drug delivery, and imparts drug resistance through various mechanisms that are still only partially characterized and understood[9][10]. In addition, the presence of high inter- and intra-tumor heterogeneity is another hallmark of PDAC [11]. Accumulating evidence indicates that morphological heterogeneity in PDAC is accompanied by a marked genetic heterogeneity[12][13]. Tumor heterogeneity results in significant variability in treatment outcomes for existing therapeutics and makes the development of new therapeutics challenging.
Because the existing treatment modalities beyond surgical resection seem to be largely ineffective in PDAC, several different aspects of pancreatic tumor biology are being investigated to identify new therapeutic strategies and targets. One of these aspects is the altered metabolism that prevails in cancers. Metabolic alterations within a tumor have long been recognized as a hallmark of cancer in general, however, the exact underlying mechanisms are only recently beginning to be uncovered[14][15][16][17]. Growing evidence indicates that metabolism is extensively reprogrammed in PDAC, both at local and systemic levels, and stroma plays a key role in this process[18][19][20]. To date, the nature of the altered metabolism in PDAC, its key drivers, and the overall impact on chemosensitivity remains unclear.
During the last two decades, pancreatic stellate cells (PSCs), also referred to as cancer-associated fibroblasts (CAFs)—the largest cellular component of PDAC tissue—have been well characterized, although important roles are still incompletely understood. PSCs interact closely with the proper malignant epithelial pancreatic cancer cells (PC cells) and with other cell types of the stroma, resulting in a growth permissive environment for pancreatic tumors[21]. Recently, PSCs have also been shown to act as a nutrient supplier to tumor cells and help them survive and proliferate in a challenging PDAC environment[14][22]. Moreover, PSCs are also implicated in the development of therapeutic resistance of PDAC[23][24].
2. Pancreatic Stellate Cells—A Key Stromal Component with Several Unknowns
In contrast to early PDAC research typically concentrated on the proper malignant epithelial cancer cells, another major stakeholder, i.e., the PSCs and CAFs, only gained significant attention in recent years[2566]. PSCs, the key pro-fibrogenic cells of the pancreas, are considered to play a crucial role in the pathobiology of various pancreatic disorders including pancreatitis and PDAC. The precise origin of PSCs remains unclear; however, they are suggested to be derived from endodermal, mesenchymal, and neuroectodermal origins[21][2667]. PSCs can be identified according to their expression of desmin, glial fibrillary acidic protein, vimentin, nestin, and neuroectodermal markers such as nerve growth factor[2667].
In the healthy pancreas, resident fibroblasts and quiescent PSCs co-exist to maintain normal gland connective tissue architecture. Quiescent PSCs transform into the activated form in response to injury or tissue damage such as chronic pancreatitis and carcinogenesis[2768]. Activated PSCs appear myofibroblast-like, express a high level of α-smooth muscle actin (α-SMA) and lose cytoplasmic lipid droplets containing anti-fibrogenic vitamin A[2869][2971][3070]. Activated PSCs are the primary cellular source of CAFs, and are considered to arise from quiescent PSCs and fibroblasts, through epithelial–mesenchymal transition (EMT), or from bone marrow-derived cells[3172]. Interestingly, with repeated or sustained injury, PSCs attain a permanently activated state that is maintained even upon the removal of the primary trigger. Activation of PSCs can occur by both autocrine and paracrine mechanisms. The sustained activation of PSCs is eventually responsible for the development of pathological fibrosis that is often irreversible[2667].
Since its first successful isolation in 1998, the role of PSCs in PDAC progression has been extensively characterized, although important aspects are still incompletely understood. Several studies have highlighted the existence of functional heterogeneity among PSCs, consistent with the notion of different subpopulations that can influence tumor progression individually or synergistically, as reviewed elsewhere[3273]. The precise nature and the functional landscape of PSCs are not yet fully known. Some of the unresolved questions regarding the nature of PSCs include their exact origin, the transition from quiescent to activated form, and differences between activated PSCs and CAFs. Interestingly, CAFs demonstrate high transcriptional and functional inter- and intra-tumoral heterogeneity, as distinct subpopulations are reported to produce complex effects on the growth and progression of PDAC and on PDAC therapy responses[3374][3475][3576][3677][3778][3879]. Öhlund et al.[3374] demonstrated two spatially and functionally distinct CAF subtypes—myofibroblastic (myCAFs) and inflammatory (iCAFs). The myCAFs exhibited high α-SMA expression and were most prevalent close to tumor foci as opposed to the iCAFs, which were located more distantly from the neoplastic cells within the dense stroma and expressed low α-SMA and high inflammatory mediators and chemokines. Transcription profiling defined a contractile and stroma remodeling phenotype for myCAFs and a secretory phenotype for iCAFs[3374]. As a matter of fact, CAFs and activated PSCs are different stromal cell populations in PDAC. Although both cell types share common biomarkers, none of these are specific[3374][3980]. A clear distinction between CAFs and activated PSCs is still under debate. Several studies over the years revealed a broad functional profile of PSCs by which they contribute to overall tumor progression. However, various attempts to alter or target PSCs and their secretions have largely been unsuccessful[4081], underscoring the importance of future investigations into the roles of PSCs in attempts to develop new and effective treatment regimens for PDAC.
Following the failure of stromal-depletion approaches, the interactions between tumor cells and different stromal components, particularly PSCs, and their multifaceted contributions to PDAC progression have been thoroughly re-assessed[21][2566][4182]. Among several PSC-induced changes in cancer cells, metabolic reprogramming conducive to tumor growth and reduced chemoresponse are two major changes that are integral to tumor progression (Figure 1). In fact, two fundamental questions have gained particular attention recently, which are (i) how do PSCs help tumor cells to survive and proliferate in a hypoxic, nutrient-poor metabolically challenging environment in PDAC and (ii) to what extent is the poor response to existing therapeutics in PDAC a consequence of metabolic rewiring? In contrast to various studies that have shown that PSCs modulate cancer cell chemosensitivity and also contribute to therapy resistance[23][24][4283], it is only recently that PSCs are being explored for their role in metabolic regulation of PDAC, especially as a nutrient supplier[14][22][4384].
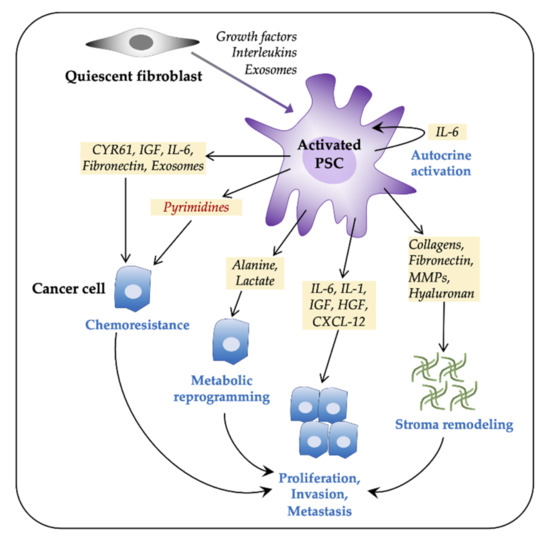
Figure 1. Pancreatic stellate cells (PSCs) facilitate tumor progression in pancreatic ductal adenocarcinoma (PDAC). Pancreatic tumors consist of a large number of activated fibroblasts, which are myofibroblast-like cells and are referred to as PSCs. Fibroblasts are activated by both paracrine and autocrine signals. Activated PSCs promote cancer cell proliferation, invasiveness, metastasis, and chemoresistance via various paracrine, exosome-mediated, or autophagic mechanisms. PSCs contribute to tumor progression by metabolic reprogramming and by extracellular matrix components-mediated stromal remodeling. CXCL12, C-X-C motif chemokine 12; CYR61, cysteine-rich angiogenic inducer 61; HGF, hepatocyte growth factor; IL, interleukin; IGF, insulin-like growth factor; MMPs, matrix metalloproteinases; PDAC, pancreatic ductal adenocarcinoma.
3.1. PSCs Facilitate Tumor Progression
Substantial evidence indicates that stroma forms a supportive niche for pancreatic tumors to grow despite adverse conditions and that PSCs in particular are a significant factor in this process[4485][4586][4687][4788]. Tumor cells recruit PSCs via mitogenic and fibrogenic factors, which promote activation, proliferation, migration, and their extracellular matrix (ECM) remodeling capability. For instance, according to Bachem et al.[4586], the supernatants from Mia PaCa-2, Panc-1, and SW850 cells stimulate PSCs proliferation and ECM synthesis in a dose-dependent manner. These effects were abrogated by neutralizing antibodies against platelet-derived growth factor (PDGF), fibroblast growth factor 2 [4889], and transforming growth factor-β1 (TGF-β1)[4990]. Moreover, cancer cells secrete ECM metalloproteinase inducer (EMMPRIN) that promotes the synthesis of matrix metalloproteinase (MMP)-2 by PSCs, which is crucial for degradation of the basement membrane and thereby influence cancer invasion and metastasis[5091]. According to Erkan et al., PSCs, once stimulated by cancer cells, remain active via an autocrine periostin loop even under active treatment, sustaining fibrogenic stellate cell activity and tumor progression[5192]. Similarly, periostin was expressed strongly in PSCs and in the stroma. Increased expression of periostin was shown to be associated with advanced disease state and reduced survival[5293].
Activated PSCs produce high levels of growth factors and inflammatory mediators that maintain their activated phenotype. In particular, inflammation functions as a promoter of tumorigenesis[5394] providing tumor cells with survival advantages through the IL-1α signaling cascade, as shown by Tjomsland et al.[5495]. PSC-conditioned medium (PSC-CM) affects cancer cell behavior including proliferation, migration, and invasion via a variety of different molecular mechanisms[4687][5596][5697][5798][5899]. For example, PSCs stimulate invasion and migration of PC cells by production of stromal cell-derived factor-1 (SDF-1) secreted protein acidic and rich in cysteine (SPARC) and matrix metalloproteinases (MMPs)[4485][5091][59100][60101][61102]. Moreover, upon activation, PSCs acquire a proinflammatory phenotype and secrete proteins in a eukaryotic translation initiation factor eIF4E-dependent manner[62103]. According to Lu et al. [63104], the PSC-secreted ECM component collagen-I is a major mediator of migration of PC cells via activation of the α2β1 integrin-FAK signaling pathway. Co-culturing of PSCs with cancer cells promotes EMT[64105], whereas in vivo co-injection of PSCs with cancer cells in an orthotopic murine model revealed increased primary tumor incidence, tumor size, and distant metastasis[4788]. Interestingly, Xu et al. [65106] suggest that human PSCs were able to accompany cancer cells to metastatic sites and stimulate angiogenesis. Human PDAC-derived PSCs were also shown to promote cell viability and invasion of Mia PaCa-2 cells in vitro and an increased rate of tumor growth was seen in mice bearing inoculated PSCs and Mia PaCa-2 cells as compared to inoculated Mia PaCa-2 alone[66107]. These findings clearly demonstrated reciprocal interactions between PSCs and cancer cells. As such, cancer cells recruit and activate PSCs, which in turn produce a conducive environment to promote local tumor growth and metastatic expansion. However, the precise molecular mechanisms underlying many of these PSC-induced effects on tumor growth and metastasis remain elusive.
3.2. PSCs and Gemcitabine Chemoresistance
The dismal clinical outcome of current PDAC chemotherapy is partially linked to chemoresistance that typically exists de novo and/or acquired during exposure of tumors to various chemotherapeutic agents[8]. Despite modest clinical effects, gemcitabine has been the preferred drug for all stages of PDAC since its inception[8][23]. Although the exact mechanisms of gemcitabine chemoresistance are only partly understood[8], biophysical properties of the tumor, in addition to pharmacokinetics and pharmacodynamics of the drug are involved[67108][68109][69110]. The cause of gemcitabine chemoresistance in PDAC is clearly multifactorial, and may be generally classified into four major mechanisms—(i) reduced intracellular transport, (ii) enhanced drug efflux, (iii) dysregulated drug metabolism, and (iv) changes in cellular signaling that negatively affect drug-induced cytotoxicity[23]. In this context, it is noteworthy that despite constituting the major cellular component of PDAC tumors, PSCs were largely ignored in the past for their putative role in the regulation of cancer cell chemosensitivity. It is only recently that PSCs were recognized to modulate tumor cell chemoresponse[23][24][5899][70111]. PSCs promote chemoresistance to gemcitabine through multiple mechanisms involving the physical barrier mechanisms of the stroma, altered drug bioavailability including a recently suggested drug scavenging effect, and PSC-induced molecular changes in tumor cells[24][4283][69110][71112][72113].
3.2.1. Stroma—Biophysical Barrier and Drug Scavenging
With the activation of PSCs, there is a consistent propagation of fibrosis, which is considered a major limiting factor in the delivery of therapeutics to the carcinoma cells. The hypovascular and dense stroma in PDAC is believed to act as a physical barrier to efficient drug delivery[72113][73114][74115][75116]. The chemotherapeutic agents are required to cross the blood vessel wall, traverse the extracellular compartment, and enter the cytoplasm of cancer cells to ultimately reach their target sites[76117]. The high interstitial fluid pressure (IFP) generally present in PDAC stroma limits the movement of cytotoxic agents from the vasculature to the extracellular compartment[73114][77118][78119]. Rice et al. reported that matrix stiffness in PDAC induced chemoresistance to paclitaxel but not to gemcitabine in vitro, suggesting that environmental rigidity may underlie some aspects of chemoresistance[79120]. PDAC stroma is often rich in the ECM component hyaluronan (hyaluronic acid; HA), which is a major contributor to increased IFP thereby limiting drug delivery to cancer cells. Consequently, targeting HA has been considered an attractive target to overcome chemoresistance. Recent studies have shown that enzymatic targeting of HA in PDAC using pegvorhyaluronidase alfa (PEGPH20), depleted stromal HA, reduced IFP, and substantially improved the effects of gemcitabine[73114][80121].
In recent years, exosomes, extracellular vesicles secreted by CAFs have been implicated in the tumor–stroma crosstalk associated with chemoresistance. Richards et al. found that CAFs exposed to gemcitabine significantly increased exosome release, which subsequently stimulated the expression of the transcription factor Snail, a known chemoresistance-inducing factor, in the recipient tumor cells, causing enhanced proliferation and drug resistance [81122]. Furthermore, a study by Fang et al.[82123] demonstrated that human CAFs-derived exosomal miRNA, miR-106b, contributes to gemcitabine resistance in PDAC. According to a recent study by Hessmann et al.[71112], PSCs entrap active gemcitabine intracellularly, causing drug scavenging and thereby limiting the availability of gemcitabine to tumor cells that may subsequently contribute to the clinical failure of gemcitabine. This finding suggests a novel and alternative mechanism underlying the functional biophysical stroma barrier for drug delivery in PDAC. However, the phenomenon of PSC-induced drug scavenging requires further validations. In contrast to the findings by Hessmann et al.[83124], our recent study in human PDAC-derived paired primary co-cultures of PSCs and cancer cells demonstrates that gemcitabine uptake and its intracellular processing in PSCs are significantly lower compared to uptake and processing capacities in primary PC cells and PDAC cell lines.
3.2.2. PSC—Tumor Cell Crosstalk in Induction of Chemoresistance
Multiple lines of evidence implicate interactions between PSCs and cancer cells in the induction of gemcitabine chemoresistance in PDAC[8][23][24][2667][4788][84125][85126]. According to Miyamoto et al., PC cells cultured on a fibronectin-coated surface showed a varying degree of increased resistance to gemcitabine, suggesting that ECM proteins are implicated in the induction of chemoresistance[86127]. Similarly, our recent study identifies PSC-secreted fibronectin as a chemoresistance-inducing factor in PC cells and that PSCs induce a varying degree of resistance to gemcitabine via activation of MAPK/ERK signaling in seven different human PC cell lines[24]. The study further observed that compared to fibronectin alone, the PSC-CM induced significantly higher chemoresistance, suggesting that other components of the PSC-secretome such as collagens may also contribute to the chemoresistance. Knowledge of the role of different collagens in chemoresistance in PDAC is limited, however, it is known that high gene expression of collagen correlated with drug resistance in for example ovarian and breast cancer cell lines[87128][88129][89130]. Panc-1 cells cultured in 3D collagen have shown increased gemcitabine resistance due to enhanced histone acetylation, possibly affecting gene expression through activation of PI3K/AKT and ERK signaling pathways, leading to increased proliferation despite drug treatment[90131][91132]. Human PSCs were shown to promote chemoresistance in Mia PaCa-2 cells by downregulation of gemcitabine-induced apoptosis[66107]. Similarly, both in vitro and in vivo gene silencing of periostin, a secretory protein exclusively expressed by PSCs, revealed that periostin regulates gemcitabine-induced apoptosis and plays a role in the progression of chemoresistance[92133].
Several growth factors and pro-inflammatory components secreted by PSCs have also been shown to induce therapeutic resistance in PDAC. For instance, CAF-secreted insulin-like growth factors (IGF)-1 and -2 promote chemoresistance to gemcitabine in PC cells, whereas pharmacologic inhibition of IGF re-sensitizes PDAC to gemcitabine in vivo[93134]. Moreover, Long et al. showed that CAF-derived interleukin (IL)-6 induces STAT3 activation in PC cells, which mediates PDAC chemoresistance[94135], whereas Duluc et al. demonstrated a role of the mTOR/4E-BP1 pathway in promoting chemoresistance via autocrine and paracrine IL-6 loop[95136]. Neumann et al. demonstrated that direct cell–cell contact and high levels of IL-6 during co-cultures of CAFs and PDAC cell lines correlate with a high degree of chemoresistance[96137]. Furthermore, PSCs contribute to PDAC chemoresistance via the release of nitric oxide, IL-1β, and type 1 collagen signaling[5192][97138][98139][99140]. According to Wei et al., CAF-secreted SDF-1 (CXCL12) stimulated malignant progression and gemcitabine resistance in PDAC are partially linked to paracrine induction of SATB-1, the special AT-rich sequence-binding protein 1[100141]. Moreover, while PSCs strongly express hepatocyte growth factor (HGF), its receptor c-Met is mainly expressed by cancer cells. Functionally, paracrine HGF from PSCs can activate the c-Met/PI3K/Akt pathway in cancer cells, leading to inhibition of cancer cell apoptosis and induction of chemoresistance to gemcitabine[101142]. Co-culture studies of PSCs and PDAC cell lines BxPC-3 and Panc-1 have demonstrated induction of gemcitabine chemoresistance via increased expression of hairy and enhancer of split-1 (HES1), which is also associated with poor prognosis of PDAC patients. The PSC-induced chemoresistance was reversed by HES 1 siRNA and a Notch signaling inhibitor[102143].
3.2.3. Intracellular Processing of Gemcitabine in PSCs and Cancer Cells
Gemcitabine (dFdC) is a deoxynucleoside analog that requires transmembrane transport and intracellular activation by phosphorylation to generate its active form dFdCTP that ultimately inhibits DNA synthesis and induces apoptosis [8]. The pharmacokinetics of gemcitabine in tumor cells is regulated by human equilibrative nucleoside transporters (hENTs)-mediated uptake, activation by deoxycytidine kinase (DCK), and inactivation by cytidine deaminase (CDA), deoxycytidylate deaminase (DCTD), and cytosolic 5′-nucleotidases (NT5C1A) (Figure 2). Altered expression of these enzymes may lead to reduced gemcitabine-induced cytotoxicity, and thereby chemoresistance[8]. The nucleoside transporters hENT1 and hCNT3 are crucial mediators of gemcitabine uptake in PC cells, and their downregulation causes chemoresistance[8]. According to Hesler et al.[72113], PSCs are a source of cysteine-rich angiogenic inducer 61 (CYR61), which promotes gemcitabine resistance in PC cells by downregulating nucleoside transporters hENT1 and hCNT3, subsequently reducing drug uptake. Moreover, PSC-secretomes affect cancer cell chemosensitivity. For example, the conditioned medium from PSCs and other fibroblasts protects PC cells from gemcitabine-induced cytotoxicity because PSCs secrete deoxycytidine, which competes with gemcitabine processing by DCK in PC cells, thereby promoting chemoresistance[4283].
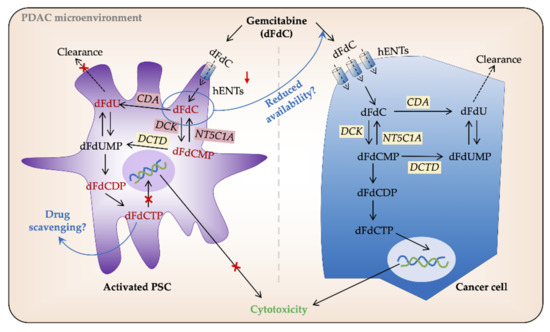
Figure 2. Gemcitabine processing in PSCs and cancer cells. In pancreatic cancer (PC) cells gemcitabine (2′,2′-difluoro-2′-deoxycytidine; dFdC) is transported intracellularly by human equilibrative nucleoside transporters (hENTs). Once inside the cell, its major proportion is inactivated by cytidine deaminase (CDA) and secreted extracellularly. The remaining gemcitabine is activated, mainly by deoxycytidine kinase (DCK), to its subsequent mono-, di-, and tri-phosphate forms dFdCMP, dFdCDP, and dFdCTP, respectively. dFdCTP competes with the substrate for DNA synthesis and induces cell death by apoptosis. Low/no expression of hENTs and other gemcitabine metabolizing enzymes reduce gemcitabine uptake and processing in PSCs. However, a large volume of PDAC tumors is made up of PSCs, and thus, any amount of gemcitabine uptake by PSCs may result in its reduced availability for PC cells. Similarly, intracellular entrapment of dFdCTP by PSCs is speculated to result in drug scavenging. DCTD, deoxycytidylate deaminase; dFdU, 2′,2′-difluoro-2′-deoxyuridine; dFdUMP, 2′,2′-difluoro-2´-deoxyuridine monophosphate; NT5C1A, 5′-nucleotidase cytosolic 1A; PSC, pancreatic stellate cell.
Gemcitabine inactivating enzyme NT5C1A negatively regulates gemcitabine activity. Interestingly, murine PSCs displayed high intracellular gemcitabine levels both in vitro and in vivo, which was supported by lower levels of NT5C1A[71112]. Similarly, the same group recently showed that NT5C1A is overexpressed in both murine and human PDAC and that in mice, NT5C1A mediates gemcitabine resistance by decreasing the amounts of intracellular dFdCTP[103144]. In addition, a co-culture experiment with conditioned media from NT5C1A expressing PSCs improved gemcitabine efficacy in tumor cells[103144]. Our recent study demonstrated high and variable NT5C1A expression in PDAC cell lines including BxPC-3, Mia PaCa-2, and Panc-1, and in human PDAC-derived primary PC cells, whereas NT5C1A expression was undetectable in PSCs derived from the same tumors. Of note, NT5C1A expression did not correlate with gemcitabine chemosensitivity in this model system as the PC cells were far more chemosensitive than PSCs despite high NT5C1A expression[83124]. NT5C1A could thus offer a potential therapeutic target for PDAC treatment, however, further investigations to this end are clearly warranted. Moreover, PSCs are intrinsically resistant to the cytotoxic actions of gemcitabine. This finding could at least partially be explained by the very low or non-existent protein expression of hENT1 and DCK in human PDAC-derived primary PSCs as compared with PC cells [83124]. Lack of hENT1 and DCK causes failure of transport and intracellular activation of gemcitabine, thus causing the observed chemoresistance in PSCs. Our study further revealed a negative correlation between gemcitabine sensitivity in terms of IC50 values and the intracellular levels of active metabolites of gemcitabine, dFdCDP, and dFdCTP, both individually and in combination[83124]. Moreover, our data demonstrate that in the cancer cells there is a strong correlation between gemcitabine sensitivity and the protein expression ratio hENT1 × DCK/CDA × DCTD [83124]. This ratio could thus serve as a novel, informative and predictive marker for gemcitabine sensitivity in PDAC. Altogether these findings suggest that PSCs affect PDAC chemoresistance in multiple and complex ways that are yet to be fully explained.
3.3. PSCs Facilitate Metabolic Rewiring in PDAC
The environmental stress within PDAC imposes nutrient shortage in cancer cells. Despite diverse mechanisms promoting extracellular glucose acquisition in cancer cells via the Warburg or the reverse Warburg effect, enhanced glucose metabolism alone cannot completely compensate for the increased energetic and biosynthetic demands of tumor cells[15][10430][10549][106145][107146]. Metabolic rewiring in cancer cells and stromal components of PDAC enables access to the recycling of nutritional substrates and alternate fuel sources, in order to sustain tumor growth and survival [18][4384]. A detailed understanding of the PSC-tumor interplay may therefore offer new targets for future treatment strategies (Figure 3).
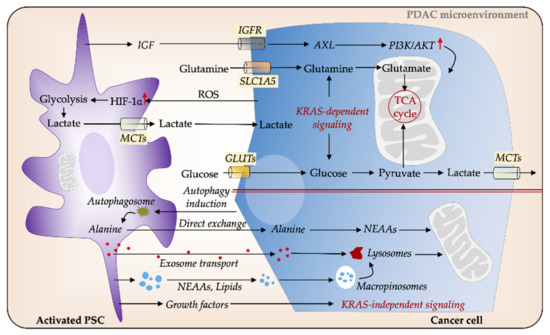
Figure 3. PSCs facilitate metabolic reprogramming in PDAC. Cancer cells secrete reactive oxygen species (ROS), which induces glycolysis in PSCs, thereby increasing the release of nutrients such as lactate, which in turn feed the energy demands of cancer cells. Several cytokines and signaling pathways mediate metabolic interactions between PSCs and PC cells via KRAS-dependent pathways. Insulin-like growth factor (IGF) increases mitochondrial respiration in cancer cells via IGFR/AXL axis. Glucose and glutamine are major sources of carbon for cancer cells. PSCs-secreted non-essential amino acids (NEAAs), such as autophagy-induced alanine, can serve as an alternative energy source to fuel cancer cells. Cancer cells can obtain nutrients from extracellular proteins for supporting their growth through upregulated macropinocytosis. In addition, PSCs-derived growth factors (GFs) and exosomes play a pivotal role in cancer cell metabolic balance. GLUT, glucose transporter; MCT; monocarboxylate transporter; PDAC, pancreatic ductal adenocarcinoma; PSC, pancreatic stellate cell.
PSCs are postulated to be the alternate energy supplier for tumor cells in PDAC. PSCs are to a significant degree able to reprogram the metabolic machinery of PDAC, in particular, the metabolic crosstalk between PSCs and tumor cells that facilitates tumor progression and invasiveness[14][22][108147]. Conversely, PC cells could force PSCs to provide the cancer cells with energy and nutrients. PSCs surrounding tumor cells also undergo a metabolic transition into a phenotype that displays characteristics corresponding to the Warburg effect. Compared to quiescent fibroblasts, activated PSCs take up more glucose and produce more lactate, which subsequently is actively taken up by tumor cells to support their growth[109148]. PSC-derived non-essential amino acids (NEAAs) also provide nutrients to feed PC cells.
Recently, PSCs were shown to regulate tumor cell metabolic activities through autophagic alanine secretion[22]. Autophagy is also required for the activation of PSCs, in addition to being associated with the growth and progression of pancreatic tumors in mice[110149]. Tumor cells increase autophagy in PSCs via unknown mechanisms and stimulate alanine secretion, which serves as an alternative carbon source of pancreatic tumors. The PSC-secreted alanine is taken up by tumor cells, thus outcompeting glucose and glutamine to support the tricarboxylic acid (TCA) cycle in order to produce NEAAs and lipids[14][22]. Furthermore, PSC-derived exosomes provide another source of alanine to fuel cancer cells and the resulting metabolic remodeling was described to be independent of KRAS mutation[111150]. Collectively, the tumor–stromal metabolic crosstalk directs PSCs to supply energy and assist tumor cells to thrive in the hostile, hypoxic, and nutrient-poor PDAC environment.
Hypoxia-inducible factor (HIF)-1α, a protein expressed under hypoxic conditions, is overexpressed in PDAC and associated with poor prognosis[112151][113152][114153][115154]. Hypoxia increases survival, proliferation, EMT, and invasiveness of PC cells and promotes resistance to therapy through HIF-1α -dependent and -independent mechanisms [116155][117156][118157][119158][120159]. Moreover, tumor cells stabilize HIF-1α in PSCs by increasing ROS production to increase glycolysis, leading to the formation of a “pseudo-hypoxic” environment for PSCs[121160]. The metabolic interplay between PSCs and cancer cells is considered a consequence of genetic mutations combined with a comprehensive paracrine signaling network [14][15][122161]. Oncogenic KRAS signaling has been reported to play a predominant role in multiple aspects of PDAC metabolism, including adaptive metabolic responses and PSC-tumor cell interplay[14][123162][124163]. It is becoming increasingly apparent that KRAS mutations manipulate signaling in both tumor cells and neighboring PSCs and influence the metabolic interactions between the two cell types[125164]. KRAS promotes sonic hedgehog secretion from PC cells, which causes activation of PSCs to induce secretion of various cytokines such as IGF1, GAS6, and GM-CSF. Subsequently, PSCs send reciprocal feedback signals to PC cells via the IGF1R/AXL axis, which activates downstream PI3K-AKT phosphorylation. This leads to increased mitochondrial respiratory capacity in PC cells and subsequently elevated oxygen availability under hypoxia[125164].
References
- Kamisawa, T.; Wood, L.D.; Itoi, T.; Takaori, K. Pancreatic cancer. Lancet 2016, 388, 73–85.
- Rahib, L.; Smith, B.D.; Aizenberg, R.; Rosenzweig, A.B.; Fleshman, J.M.; Matrisian, L.M. Projecting cancer incidence and deaths to 2030: The unexpected burden of thyroid, liver, and pancreas cancers in the united states. Cancer Res. 2014, 74, 2913–2921.
- Kleeff, J.; Korc, M.; Apte, M.; La Vecchia, C.; Johnson, C.D.; Biankin, A.V.; Neale, R.E.; Tempero, M.; Tuveson, D.A.; Hruban, R.H.; et al. Pancreatic cancer. Nat. Rev. Dis. Primers 2016, 2, 16022.
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2019. CA Cancer J. Clin. 2019, 69, 7–34.
- Adamska, A.; Domenichini, A.; Falasca, M. Pancreatic ductal adenocarcinoma: Current and evolving therapies. Int. J. Mol. Sci. 2017, 18, 1338.
- Hajatdoost, L.; Sedaghat, K.; Walker, E.J.; Thomas, J.; Kosari, S. Chemotherapy in pancreatic cancer: A systematic review. Medicina 2018, 54, 48.
- Burris, H.A.; Moore, M.J.; Andersen, J.; Green, M.R.; Rothenberg, M.L.; Modiano, M.R.; Cripps, M.C.; Portenoy, R.K.; Storniolo, A.M.; Tarassoff, P.; et al. Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: A randomized trial. J. Clin. Oncol. 1997, 15, 2403–2413.
- Amrutkar, M.; Gladhaug, I.P. Pancreatic cancer chemoresistance to gemcitabine. Cancers 2017, 9, 157.
- Neesse, A.; Michl, P.; Frese, K.K.; Feig, C.; Cook, N.; Jacobetz, M.A.; Lolkema, M.P.; Buchholz, M.; Olive, K.P.; Gress, T.M.; et al. Stromal biology and therapy in pancreatic cancer. Gut 2011, 60, 861–868.
- Neesse, A.; Bauer, C.A.; Öhlund, D.; Lauth, M.; Buchholz, M.; Michl, P.; Tuveson, D.A.; Gress, T.M. Stromal biology and therapy in pancreatic cancer: Ready for clinical translation? Gut 2019, 68, 159–171.
- Orth, M.; Metzger, P.; Gerum, S.; Mayerle, J.; Schneider, G.; Belka, C.; Schnurr, M.; Lauber, K. Pancreatic ductal adenocarcinoma: Biological hallmarks, current status, and future perspectives of combined modality treatment approaches. Radiat. Oncol. 2019, 14, 141.
- Verbeke, C. Morphological heterogeneity in ductal adenocarcinoma of the pancreas—Does it matter? Pancreatology 2016, 16, 295–301.
- Biankin, A.V.; Waddell, N.; Kassahn, K.S.; Gingras, M.C.; Muthuswamy, L.B.; Johns, A.L.; Miller, D.K.; Wilson, P.J.; Patch, A.M.; Wu, J.; et al. Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nature 2012, 491, 399–405.
- Halbrook, C.J.; Lyssiotis, C.A. Employing metabolism to improve the diagnosis and treatment of pancreatic cancer. Cancer Cell 2017, 31, 5–19.
- Perera, R.M.; Bardeesy, N. Pancreatic cancer metabolism: Breaking it down to build it back up. Cancer Discov. 2015, 5, 1247–1261.
- Vernucci, E.; Abrego, J.; Gunda, V.; Shukla, S.K.; Dasgupta, A.; Rai, V.; Chaika, N.; Buettner, K.; Illies, A.; Yu, F.; et al. Metabolic alterations in pancreatic cancer progression. Cancers 2019, 12, 2.
- Hardie, R.A.; van Dam, E.; Cowley, M.; Han, T.L.; Balaban, S.; Pajic, M.; Pinese, M.; Iconomou, M.; Shearer, R.F.; McKenna, J.; et al. Mitochondrial mutations and metabolic adaptation in pancreatic cancer. Cancer Metab. 2017, 5, 2.
- Vaziri-Gohar, A.; Zarei, M.; Brody, J.R.; Winter, J.M. Metabolic dependencies in pancreatic cancer. Front. Oncol. 2018, 8, 617.
- Olivares, O.; Vasseur, S. Metabolic rewiring of pancreatic ductal adenocarcinoma: New routes to follow within the maze. Int. J. Cancer 2016, 138, 787–796.
- Knudsen, E.S.; Balaji, U.; Freinkman, E.; McCue, P.; Witkiewicz, A.K. Unique metabolic features of pancreatic cancer stroma: Relevance to the tumor compartment, prognosis, and invasive potential. Oncotarget 2016, 7, 78396–78411.
- Pothula, S.P.; Pirola, R.C.; Wilson, J.S.; Apte, M.V. Pancreatic stellate cells: Aiding and abetting pancreatic cancer progression. Pancreatology 2020, 20, 409–418.
- Sousa, C.M.; Biancur, D.E.; Wang, X.; Halbrook, C.J.; Sherman, M.H.; Zhang, L.; Kremer, D.; Hwang, R.F.; Witkiewicz, A.K.; Ying, H.; et al. Pancreatic stellate cells support tumour metabolism through autophagic alanine secretion. Nature 2016, 536, 479–483.
- McCarroll, J.A.; Naim, S.; Sharbeen, G.; Russia, N.; Lee, J.; Kavallaris, M.; Goldstein, D.; Phillips, P.A. Role of pancreatic stellate cells in chemoresistance in pancreatic cancer. Front. Physiol. 2014, 5, 141.
- Amrutkar, M.; Aasrum, M.; Verbeke, C.S.; Gladhaug, I.P. Secretion of fibronectin by human pancreatic stellate cells promotes chemoresistance to gemcitabine in pancreatic cancer cells. BMC Cancer 2019, 19, 596.
- Thomas, D.; Radhakrishnan, P. Pancreatic stellate cells: The key orchestrator of the pancreatic tumor microenvironment. Adv. Exp. Med. Biol. 2020, 1234, 57–70.DeBerardinis, R.J.; Chandel, N.S. Fundamentals of cancer metabolism. Sci. Adv. 2016, 2, e1600200.
- Wilson, J.S.; Pirola, R.C.; Apte, M.V. Stars and stripes in pancreatic cancer: Role of stellate cells and stroma in cancer progression. Front. Physiol. 2014, 5, 52.Vazquez, A.; Kamphorst, J.J.; Markert, E.K.; Schug, Z.T.; Tardito, S.; Gottlieb, E. Cancer metabolism at a glance. J. Cell Sci. 2016, 129, 3367–3373.
- Phillips, P. Pancreatic stellate cells and fibrosis. In Pancreatic Cancer and Tumor Microenvironment; Grippo, P.J., Munshi, H.G., Eds.; Transworld Research Network: Trivandrum, India, 2012.Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314.
- Bachem, M.G.; Schneider, E.; Gross, H.; Weidenbach, H.; Schmid, R.M.; Menke, A.; Siech, M.; Beger, H.; Grunert, A.; Adler, G. Identification, culture, and characterization of pancreatic stellate cells in rats and humans. Gastroenterology 1998, 115, 421–432.Lunt, S.Y.; Vander Heiden, M.G. Aerobic glycolysis: Meeting the metabolic requirements of cell proliferation. Annu. Rev. Cell Dev. Biol. 2011, 27, 441–464.
- Apte, M.V.; Haber, P.S.; Applegate, T.L.; Norton, I.D.; McCaughan, G.W.; Korsten, M.A.; Pirola, R.C.; Wilson, J.S. Periacinar stellate shaped cells in rat pancreas: Identification, isolation, and culture. Gut 1998, 43, 128–133.Koppenol, W.H.; Bounds, P.L.; Dang, C.V. Otto warburg’s contributions to current concepts of cancer metabolism. Nat. Rev. Cancer 2011, 11, 325–337.
- Apte, M.V.; Haber, P.S.; Darby, S.J.; Rodgers, S.C.; McCaughan, G.W.; Korsten, M.A.; Pirola, R.C.; Wilson, J.S. Pancreatic stellate cells are activated by proinflammatory cytokines: Implications for pancreatic fibrogenesis. Gut 1999, 44, 534–541.Fu, Y.; Liu, S.; Yin, S.; Niu, W.; Xiong, W.; Tan, M.; Li, G.; Zhou, M. The reverse warburg effect is likely to be an achilles’ heel of cancer that can be exploited for cancer therapy. Oncotarget 2017, 8, 57813–57825.
- Rasheed, Z.A.; Matsui, W.; Maitra, A. Pathology of pancreatic stroma in pdac. In Pancreatic Cancer and Tumor Microenvironment; Grippo, P.J., Munshi, H.G., Eds.; Transworld Research Network: Trivandrum, India, 2012.Danhier, P.; Bański, P.; Payen, V.L.; Grasso, D.; Ippolito, L.; Sonveaux, P.; Porporato, P.E. Cancer metabolism in space and time: Beyond the warburg effect. Biochim. Biophys. Acta Bioenerg. 2017, 1858, 556–572.
- Xue, R.; Jia, K.; Wang, J.; Yang, L.; Wang, Y.; Gao, L.; Hao, J. A rising star in pancreatic diseases: Pancreatic stellate cells. Front. Physiol. 2018, 9, 754.Wallace, D.C. Mitochondria and cancer. Nat. Rev. Cancer 2012, 12, 685–698.
- Öhlund, D.; Handly-Santana, A.; Biffi, G.; Elyada, E.; Almeida, A.S.; Ponz-Sarvise, M.; Corbo, V.; Oni, T.E.; Hearn, S.A.; Lee, E.J.; et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J. Exp. Med. 2017, 214, 579–596.Daemen, A.; Peterson, D.; Sahu, N.; McCord, R.; Du, X.; Liu, B.; Kowanetz, K.; Hong, R.; Moffat, J.; Gao, M.; et al. Metabolite profiling stratifies pancreatic ductal adenocarcinomas into subtypes with distinct sensitivities to metabolic inhibitors. Proc. Natl. Acad. Sci. USA 2015, 112, E4410–E4417.
- Neuzillet, C.; Tijeras-Raballand, A.; Ragulan, C.; Cros, J.; Patil, Y.; Martinet, M.; Erkan, M.; Kleeff, J.; Wilson, J.; Apte, M.; et al. Inter- and intra-tumoural heterogeneity in cancer-associated fibroblasts of human pancreatic ductal adenocarcinoma. J. Pathol. 2019, 248, 51–65.Pavlides, S.; Whitaker-Menezes, D.; Castello-Cros, R.; Flomenberg, N.; Witkiewicz, A.K.; Frank, P.G.; Casimiro, M.C.; Wang, C.; Fortina, P.; Addya, S.; et al. The reverse warburg effect: Aerobic glycolysis in cancer associated fibroblasts and the tumor stroma. Cell Cycle 2009, 8, 3984–4001.
- Yu, Y.; Schuck, K.; Friess, H.; Kong, B. Targeting aggressive fibroblasts to enhance the treatment of pancreatic cancer. Expert Opin. Ther. Targets 2020, 1–9.Xu, X.D.; Shao, S.X.; Jiang, H.P.; Cao, Y.W.; Wang, Y.H.; Yang, X.C.; Wang, Y.L.; Wang, X.S.; Niu, H.T. Warburg effect or reverse warburg effect? A review of cancer metabolism. Oncol. Res. Treat. 2015, 38, 117–122.
- Elyada, E.; Bolisetty, M.; Laise, P.; Flynn, W.F.; Courtois, E.T.; Burkhart, R.A.; Teinor, J.A.; Belleau, P.; Biffi, G.; Lucito, M.S.; et al. Cross-species single-cell analysis of pancreatic ductal adenocarcinoma reveals antigen-presenting cancer-associated fibroblasts. Cancer Discov. 2019, 9, 1102–1123.Lee, M.; Yoon, J.H. Metabolic interplay between glycolysis and mitochondrial oxidation: The reverse warburg effect and its therapeutic implication. World J. Biol. Chem. 2015, 6, 148–161.
- Pereira, B.A.; Vennin, C.; Papanicolaou, M.; Chambers, C.R.; Herrmann, D.; Morton, J.P.; Cox, T.R.; Timpson, P. Caf subpopulations: A new reservoir of stromal targets in pancreatic cancer. Trends Cancer 2019, 5, 724–741.Wilde, L.; Roche, M.; Domingo-Vidal, M.; Tanson, K.; Philp, N.; Curry, J.; Martinez-Outschoorn, U. Metabolic coupling and the reverse warburg effect in cancer: Implications for novel biomarker and anticancer agent development. Semin. Oncol. 2017, 44, 198–203.
- Helms, E.; Onate, M.K.; Sherman, M.H. Fibroblast heterogeneity in the pancreatic tumor microenvironment. Cancer Discov. 2020, 10, 648–656.Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674.
- Nielsen, M.F.; Mortensen, M.B.; Detlefsen, S. Key players in pancreatic cancer-stroma interaction: Cancer-associated fibroblasts, endothelial and inflammatory cells. World J. Gastroenterol. 2016, 22, 2678–2700.Pavlova, N.N.; Thompson, C.B. The emerging hallmarks of cancer metabolism. Cell Metab. 2016, 23, 27–47.
- Weniger, M.; Honselmann, K.C.; Liss, A.S. The extracellular matrix and pancreatic cancer: A complex relationship. Cancers 2018, 10, 316.Cohen, R.; Neuzillet, C.; Tijeras-Raballand, A.; Faivre, S.; de Gramont, A.; Raymond, E. Targeting cancer cell metabolism in pancreatic adenocarcinoma. Oncotarget 2015, 6, 16832–16847.
- Mekapogu, A.R.; Pothula, S.P.; Pirola, R.C.; Wilson, J.S.; Apte, M.V. Multifunctional role of pancreatic stellate cells in pancreatic cancer. Ann. Pancreat. Cancer 2019, 2, 10.Liu, J.; Ji, S.; Liang, C.; Qin, Y.; Jin, K.; Liang, D.; Xu, W.; Shi, S.; Zhang, B.; Liu, L.; et al. Critical role of oncogenic kras in pancreatic cancer (review). Mol. Med. Rep. 2016, 13, 4943–4949.
- Dalin, S.; Sullivan, M.R.; Lau, A.N.; Grauman-Boss, B.; Mueller, H.S.; Kreidl, E.; Fenoglio, S.; Luengo, A.; Lees, J.A.; Vander Heiden, M.G.; et al. Deoxycytidine release from pancreatic stellate cells promotes gemcitabine resistance. Cancer Res. 2019, 79, 5723–5733.Waters, A.M.; Der, C.J. Kras: The critical driver and therapeutic target for pancreatic cancer. Cold Spring Harb. Perspect. Med. 2018, 8, a031435.
- Sherman, M.H.; Yu, R.T.; Tseng, T.W.; Sousa, C.M.; Liu, S.; Truitt, M.L.; He, N.; Ding, N.; Liddle, C.; Atkins, A.R.; et al. Stromal cues regulate the pancreatic cancer epigenome and metabolome. Proc. Natl. Acad. Sci. USA 2017, 114, 1129–1134.Ying, H.; Kimmelman, A.C.; Lyssiotis, C.A.; Hua, S.; Chu, G.C.; Fletcher-Sananikone, E.; Locasale, J.W.; Son, J.; Zhang, H.; Coloff, J.L.; et al. Oncogenic kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell 2012, 149, 656–670.
- Apte, M.V.; Park, S.; Phillips, P.A.; Santucci, N.; Goldstein, D.; Kumar, R.K.; Ramm, G.A.; Buchler, M.; Friess, H.; McCarroll, J.A.; et al. Desmoplastic reaction in pancreatic cancer: Role of pancreatic stellate cells. Pancreas 2004, 29, 179–187.Santana-Codina, N.; Roeth, A.A.; Zhang, Y.; Yang, A.; Mashadova, O.; Asara, J.M.; Wang, X.; Bronson, R.T.; Lyssiotis, C.A.; Ying, H.; et al. Oncogenic kras supports pancreatic cancer through regulation of nucleotide synthesis. Nat. Commun. 2018, 9, 4945.
- Bachem, M.G.; Schünemann, M.; Ramadani, M.; Siech, M.; Beger, H.; Buck, A.; Zhou, S.; Schmid-Kotsas, A.; Adler, G. Pancreatic carcinoma cells induce fibrosis by stimulating proliferation and matrix synthesis of stellate cells. Gastroenterology 2005, 128, 907–921.Nollmann, F.I.; Ruess, D.A. Targeting mutant kras in pancreatic cancer: Futile or promising? Biomedicines 2020, 8, 281.
- Hwang, R.F.; Moore, T.; Arumugam, T.; Ramachandran, V.; Amos, K.D.; Rivera, A.; Ji, B.; Evans, D.B.; Logsdon, C.D. Cancer-associated stromal fibroblasts promote pancreatic tumor progression. Cancer Res. 2008, 68, 918–926.Kimmelman, A.C. Metabolic dependencies in ras-driven cancers. Clin. Cancer Res. 2015, 21, 1828–1834.
- Vonlaufen, A.; Joshi, S.; Qu, C.; Phillips, P.A.; Xu, Z.; Parker, N.R.; Toi, C.S.; Pirola, R.C.; Wilson, J.S.; Goldstein, D.; et al. Pancreatic stellate cells: Partners in crime with pancreatic cancer cells. Cancer Res. 2008, 68, 2085–2093.White, E. Exploiting the bad eating habits of ras-driven cancers. Genes Dev. 2013, 27, 2065–2071.
- Coleman, S.J.; Chioni, A.M.; Ghallab, M.; Anderson, R.K.; Lemoine, N.R.; Kocher, H.M.; Grose, R.P. Nuclear translocation of fgfr1 and fgf2 in pancreatic stellate cells facilitates pancreatic cancer cell invasion. EMBO Mol. Med. 2014, 6, 467–481.Shalapour, S.; Karin, M. Immunity, inflammation, and cancer: An eternal fight between good and evil. J. Clin. Investig. 2015, 125, 3347–3355.
- Bachem, M.G.; Zhou, S.; Buck, K.; Schneiderhan, W.; Siech, M. Pancreatic stellate cells—role in pancreas cancer. Langenbecks Arch. Surg. 2008, 393, 891–900.Kamphorst, J.J.; Nofal, M.; Commisso, C.; Hackett, S.R.; Lu, W.; Grabocka, E.; Vander Heiden, M.G.; Miller, G.; Drebin, J.A.; Bar-Sagi, D.; et al. Human pancreatic cancer tumors are nutrient poor and tumor cells actively scavenge extracellular protein. Cancer Res. 2015, 75, 544–553.
- Schneiderhan, W.; Diaz, F.; Fundel, M.; Zhou, S.; Siech, M.; Hasel, C.; Möller, P.; Gschwend, J.E.; Seufferlein, T.; Gress, T.; et al. Pancreatic stellate cells are an important source of mmp-2 in human pancreatic cancer and accelerate tumor progression in a murine xenograft model and cam assay. J. Cell Sci. 2007, 120, 512–519.Chauhan, V.P.; Boucher, Y.; Ferrone, C.R.; Roberge, S.; Martin, J.D.; Stylianopoulos, T.; Bardeesy, N.; DePinho, R.A.; Padera, T.P.; Munn, L.L.; et al. Compression of pancreatic tumor blood vessels by hyaluronan is caused by solid stress and not interstitial fluid pressure. Cancer Cell 2014, 26, 14–15.
- Erkan, M.; Kleeff, J.; Gorbachevski, A.; Reiser, C.; Mitkus, T.; Esposito, I.; Giese, T.; Büchler, M.W.; Giese, N.A.; Friess, H. Periostin creates a tumor-supportive microenvironment in the pancreas by sustaining fibrogenic stellate cell activity. Gastroenterology 2007, 132, 1447–1464.Vasseur, S.; Tomasini, R.; Tournaire, R.; Iovanna, J.L. Hypoxia induced tumor metabolic switch contributes to pancreatic cancer aggressiveness. Cancers 2010, 2, 2138–2152.
- Liu, Y.; Li, F.; Gao, F.; Xing, L.; Qin, P.; Liang, X.; Zhang, J.; Qiao, X.; Lin, L.; Zhao, Q.; et al. Role of microenvironmental periostin in pancreatic cancer progression. Oncotarget 2017, 8, 89552–89565.Derle, A.; De Santis, M.C.; Gozzelino, L.; Ratto, E.; Martini, M. The role of metabolic adaptation to nutrient stress in pancreatic cancer. Cell Stress 2018, 2, 332–339.
- Kalluri, R. The biology and function of fibroblasts in cancer. Nat. Rev. Cancer 2016, 16, 582–598.Blum, R.; Kloog, Y. Metabolism addiction in pancreatic cancer. Cell Death Dis. 2014, 5, e1065.
- Tjomsland, V.; Spångeus, A.; Välilä, J.; Sandström, P.; Borch, K.; Druid, H.; Falkmer, S.; Falkmer, U.; Messmer, D.; Larsson, M. Interleukin 1α sustains the expression of inflammatory factors in human pancreatic cancer microenvironment by targeting cancer-associated fibroblasts. Neoplasia 2011, 13, 664–675.Wise, D.R.; Thompson, C.B. Glutamine addiction: A new therapeutic target in cancer. Trends Biochem. Sci. 2010, 35, 427–433.
- Vonlaufen, A.; Phillips, P.A.; Xu, Z.; Goldstein, D.; Pirola, R.C.; Wilson, J.S.; Apte, M.V. Pancreatic stellate cells and pancreatic cancer cells: An unholy alliance. Cancer Res. 2008, 68, 7707–7710.Son, J.; Lyssiotis, C.A.; Ying, H.; Wang, X.; Hua, S.; Ligorio, M.; Perera, R.M.; Ferrone, C.R.; Mullarky, E.; Shyh-Chang, N.; et al. Glutamine supports pancreatic cancer growth through a kras-regulated metabolic pathway. Nature 2013, 496, 101–105.
- Gao, Z.; Wang, X.; Wu, K.; Zhao, Y.; Hu, G. Pancreatic stellate cells increase the invasion of human pancreatic cancer cells through the stromal cell-derived factor-1/cxcr4 axis. Pancreatology 2010, 10, 186–193.Banh, R.S.; Biancur, D.E.; Yamamoto, K.; Sohn, A.S.W.; Walters, B.; Kuljanin, M.; Gikandi, A.; Wang, H.; Mancias, J.D.; Schneider, R.J.; et al. Neurons release serine to support mrna translation in pancreatic cancer. Cell 2020, 183, 1202–1218.e1225.
- Tjomsland, V.; Aasrum, M.; Christoffersen, T.; Gladhaug, I.P. Functional heterogeneity in tumor-derived human pancreatic stellate cells: Differential expression of hgf and implications for mitogenic signaling and migration in pancreatic cancer cells. Oncotarget 2017, 8, 71672–71684.Badgley, M.A.; Kremer, D.M.; Maurer, H.C.; DelGiorno, K.E.; Lee, H.J.; Purohit, V.; Sagalovskiy, I.R.; Ma, A.; Kapilian, J.; Firl, C.E.M.; et al. Cysteine depletion induces pancreatic tumor ferroptosis in mice. Science 2020, 368, 85–89.
- Lenggenhager, D.; Amrutkar, M.; Sántha, P.; Aasrum, M.; Löhr, J.M.; Gladhaug, I.P.; Verbeke, C.S. Commonly used pancreatic stellate cell cultures differ phenotypically and in their interactions with pancreatic cancer cells. Cells 2019, 8, 23.Rabinowitz, J.D.; White, E. Autophagy and metabolism. Science 2010, 330, 1344–1348.
- Tjomsland, V.; Pomianowska, E.; Aasrum, M.; Sandnes, D.; Verbeke, C.S.; Gladhaug, I.P. Profile of mmp and timp expression in human pancreatic stellate cells: Regulation by il-1alpha and tgfbeta and implications for migration of pancreatic cancer cells. Neoplasia 2016, 18, 447–456.Commisso, C.; Davidson, S.M.; Soydaner-Azeloglu, R.G.; Parker, S.J.; Kamphorst, J.J.; Hackett, S.; Grabocka, E.; Nofal, M.; Drebin, J.A.; Thompson, C.B.; et al. Macropinocytosis of protein is an amino acid supply route in ras-transformed cells. Nature 2013, 497, 633–637.
- Koshiba, T.; Hosotani, R.; Miyamoto, Y.; Ida, J.; Tsuji, S.; Nakajima, S.; Kawaguchi, M.; Kobayashi, H.; Doi, R.; Hori, T.; et al. Expression of stromal cell-derived factor 1 and cxcr4 ligand receptor system in pancreatic cancer: A possible role for tumor progression. Clin. Cancer Res. 2000, 6, 3530–3535.Yang, S.; Wang, X.; Contino, G.; Liesa, M.; Sahin, E.; Ying, H.; Bause, A.; Li, Y.; Stommel, J.M.; Dell’antonio, G.; et al. Pancreatic cancers require autophagy for tumor growth. Genes Dev. 2011, 25, 717–729.
- Mantoni, T.S.; Schendel, R.R.; Rödel, F.; Niedobitek, G.; Al-Assar, O.; Masamune, A.; Brunner, T.B. Stromal sparc expression and patient survival after chemoradiation for non-resectable pancreatic adenocarcinoma. Cancer Biol. Ther. 2008, 7, 1806–1815.Guo, J.Y.; Chen, H.Y.; Mathew, R.; Fan, J.; Strohecker, A.M.; Karsli-Uzunbas, G.; Kamphorst, J.J.; Chen, G.; Lemons, J.M.; Karantza, V.; et al. Activated ras requires autophagy to maintain oxidative metabolism and tumorigenesis. Genes Dev. 2011, 25, 460–470.
- Marzoq, A.J.; Mustafa, S.A.; Heidrich, L.; Hoheisel, J.D.; Alhamdani, M.S.S. Impact of the secretome of activated pancreatic stellate cells on growth and differentiation of pancreatic tumour cells. Sci. Rep. 2019, 9, 5303.Davidson, S.M.; Jonas, O.; Keibler, M.A.; Hou, H.W.; Luengo, A.; Mayers, J.R.; Wyckoff, J.; Del Rosario, A.M.; Whitman, M.; Chin, C.R.; et al. Direct evidence for cancer-cell-autonomous extracellular protein catabolism in pancreatic tumors. Nat. Med. 2017, 23, 235–241.
- Lu, J.; Zhou, S.; Siech, M.; Habisch, H.; Seufferlein, T.; Bachem, M.G. Pancreatic stellate cells promote hapto-migration of cancer cells through collagen i-mediated signalling pathway. Br. J. Cancer 2014, 110, 409–420.Kallunki, T.; Olsen, O.D.; Jäättelä, M. Cancer-associated lysosomal changes: Friends or foes? Oncogene 2013, 32, 1995–2004.
- Kikuta, K.; Masamune, A.; Watanabe, T.; Ariga, H.; Itoh, H.; Hamada, S.; Satoh, K.; Egawa, S.; Unno, M.; Shimosegawa, T. Pancreatic stellate cells promote epithelial-mesenchymal transition in pancreatic cancer cells. Biochem. Biophys. Res. Commun. 2010, 403, 380–384.Perera, R.M.; Stoykova, S.; Nicolay, B.N.; Ross, K.N.; Fitamant, J.; Boukhali, M.; Lengrand, J.; Deshpande, V.; Selig, M.K.; Ferrone, C.R.; et al. Transcriptional control of autophagy-lysosome function drives pancreatic cancer metabolism. Nature 2015, 524, 361–365.
- Xu, Z.; Vonlaufen, A.; Phillips, P.A.; Fiala-Beer, E.; Zhang, X.; Yang, L.; Biankin, A.V.; Goldstein, D.; Pirola, R.C.; Wilson, J.S.; et al. Role of pancreatic stellate cells in pancreatic cancer metastasis. Am. J. Pathol. 2010, 177, 2585–2596.Lawrence, R.E.; Zoncu, R. The lysosome as a cellular centre for signalling, metabolism and quality control. Nat. Cell Biol. 2019, 21, 133–142.
- Liu, S.L.; Cao, S.G.; Li, Y.; Sun, B.; Chen, D.; Wang, D.S.; Zhou, Y.B. Pancreatic stellate cells facilitate pancreatic cancer cell viability and invasion. Oncol. Lett. 2019, 17, 2057–2062.Thomas, D.; Radhakrishnan, P. Pancreatic stellate cells: The key orchestrator of the pancreatic tumor microenvironment. Adv. Exp. Med. Biol. 2020, 1234, 57–70.
- Swayden, M.; Iovanna, J.; Soubeyran, P. Pancreatic cancer chemo-resistance is driven by tumor phenotype rather than tumor genotype. Heliyon 2018, 4, e01055.Wilson, J.S.; Pirola, R.C.; Apte, M.V. Stars and stripes in pancreatic cancer: Role of stellate cells and stroma in cancer progression. Front. Physiol. 2014, 5, 52.
- Gnanamony, M.; Gondi, C.S. Chemoresistance in pancreatic cancer: Emerging concepts. Oncol. Lett. 2017, 13, 2507–2513.Phillips, P. Pancreatic stellate cells and fibrosis. In Pancreatic Cancer and Tumor Microenvironment; Grippo, P.J., Munshi, H.G., Eds.; Transworld Research Network: Trivandrum, India, 2012.
- Adamska, A.; Elaskalani, O.; Emmanouilidi, A.; Kim, M.; Abdol Razak, N.B.; Metharom, P.; Falasca, M. Molecular and cellular mechanisms of chemoresistance in pancreatic cancer. Adv. Biol. Regul. 2018, 68, 77–87.Bachem, M.G.; Schneider, E.; Gross, H.; Weidenbach, H.; Schmid, R.M.; Menke, A.; Siech, M.; Beger, H.; Grunert, A.; Adler, G. Identification, culture, and characterization of pancreatic stellate cells in rats and humans. Gastroenterology 1998, 115, 421–432.
- Che, P.P.; Gregori, A.; Firuzi, O.; Dahele, M.; Sminia, P.; Peters, G.J.; Giovannetti, E. Pancreatic cancer resistance conferred by stellate cells: Looking for new preclinical models. Exp. Hematol. Oncol. 2020, 9, 18.Apte, M.V.; Haber, P.S.; Darby, S.J.; Rodgers, S.C.; McCaughan, G.W.; Korsten, M.A.; Pirola, R.C.; Wilson, J.S. Pancreatic stellate cells are activated by proinflammatory cytokines: Implications for pancreatic fibrogenesis. Gut 1999, 44, 534–541.
- Hessmann, E.; Patzak, M.S.; Klein, L.; Chen, N.; Kari, V.; Ramu, I.; Bapiro, T.E.; Frese, K.K.; Gopinathan, A.; Richards, F.M.; et al. Fibroblast drug scavenging increases intratumoural gemcitabine accumulation in murine pancreas cancer. Gut 2018, 67, 497–507.Apte, M.V.; Haber, P.S.; Applegate, T.L.; Norton, I.D.; McCaughan, G.W.; Korsten, M.A.; Pirola, R.C.; Wilson, J.S. Periacinar stellate shaped cells in rat pancreas: Identification, isolation, and culture. Gut 1998, 43, 128–133.
- Hesler, R.A.; Huang, J.J.; Starr, M.D.; Treboschi, V.M.; Bernanke, A.G.; Nixon, A.B.; McCall, S.J.; White, R.R.; Blobe, G.C. Tgf-beta-induced stromal cyr61 promotes resistance to gemcitabine in pancreatic ductal adenocarcinoma through downregulation of the nucleoside transporters hent1 and hcnt3. Carcinogenesis 2016, 37, 1041–1051.Rasheed, Z.A.; Matsui, W.; Maitra, A. Pathology of pancreatic stroma in pdac. In Pancreatic Cancer and Tumor Microenvironment; Grippo, P.J., Munshi, H.G., Eds.; Transworld Research Network: Trivandrum, India, 2012.
- Provenzano, P.P.; Cuevas, C.; Chang, A.E.; Goel, V.K.; Von Hoff, D.D.; Hingorani, S.R. Enzymatic targeting of the stroma ablates physical barriers to treatment of pancreatic ductal adenocarcinoma. Cancer Cell 2012, 21, 418–429.Xue, R.; Jia, K.; Wang, J.; Yang, L.; Wang, Y.; Gao, L.; Hao, J. A rising star in pancreatic diseases: Pancreatic stellate cells. Front. Physiol. 2018, 9, 754.
- Neesse, A.; Algul, H.; Tuveson, D.A.; Gress, T.M. Stromal biology and therapy in pancreatic cancer: A changing paradigm. Gut 2015, 64, 1476–1484.Öhlund, D.; Handly-Santana, A.; Biffi, G.; Elyada, E.; Almeida, A.S.; Ponz-Sarvise, M.; Corbo, V.; Oni, T.E.; Hearn, S.A.; Lee, E.J.; et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J. Exp. Med. 2017, 214, 579–596.
- Pries, A.R.; Höpfner, M.; le Noble, F.; Dewhirst, M.W.; Secomb, T.W. The shunt problem: Control of functional shunting in normal and tumour vasculature. Nat. Rev. Cancer 2010, 10, 587–593.Neuzillet, C.; Tijeras-Raballand, A.; Ragulan, C.; Cros, J.; Patil, Y.; Martinet, M.; Erkan, M.; Kleeff, J.; Wilson, J.; Apte, M.; et al. Inter- and intra-tumoural heterogeneity in cancer-associated fibroblasts of human pancreatic ductal adenocarcinoma. J. Pathol. 2019, 248, 51–65.
- Koay, E.J.; Truty, M.J.; Cristini, V.; Thomas, R.M.; Chen, R.; Chatterjee, D.; Kang, Y.; Bhosale, P.R.; Tamm, E.P.; Crane, C.H.; et al. Transport properties of pancreatic cancer describe gemcitabine delivery and response. J. Clin. Investig. 2014, 124, 1525–1536.Yu, Y.; Schuck, K.; Friess, H.; Kong, B. Targeting aggressive fibroblasts to enhance the treatment of pancreatic cancer. Expert Opin. Ther. Targets 2020, 1–9.
- Stylianopoulos, T.; Martin, J.D.; Chauhan, V.P.; Jain, S.R.; Diop-Frimpong, B.; Bardeesy, N.; Smith, B.L.; Ferrone, C.R.; Hornicek, F.J.; Boucher, Y.; et al. Causes, consequences, and remedies for growth-induced solid stress in murine and human tumors. Proc. Natl. Acad. Sci. USA 2012, 109, 15101–15108.Elyada, E.; Bolisetty, M.; Laise, P.; Flynn, W.F.; Courtois, E.T.; Burkhart, R.A.; Teinor, J.A.; Belleau, P.; Biffi, G.; Lucito, M.S.; et al. Cross-species single-cell analysis of pancreatic ductal adenocarcinoma reveals antigen-presenting cancer-associated fibroblasts. Cancer Discov. 2019, 9, 1102–1123.
- DuFort, C.C.; DelGiorno, K.E.; Carlson, M.A.; Osgood, R.J.; Zhao, C.; Huang, Z.; Thompson, C.B.; Connor, R.J.; Thanos, C.D.; Scott Brockenbrough, J.; et al. Interstitial pressure in pancreatic ductal adenocarcinoma is dominated by a gel-fluid phase. Biophys. J. 2016, 110, 2106–2119.Pereira, B.A.; Vennin, C.; Papanicolaou, M.; Chambers, C.R.; Herrmann, D.; Morton, J.P.; Cox, T.R.; Timpson, P. Caf subpopulations: A new reservoir of stromal targets in pancreatic cancer. Trends Cancer 2019, 5, 724–741.
- Rice, A.J.; Cortes, E.; Lachowski, D.; Cheung, B.C.H.; Karim, S.A.; Morton, J.P.; Del Río Hernández, A. Matrix stiffness induces epithelial-mesenchymal transition and promotes chemoresistance in pancreatic cancer cells. Oncogenesis 2017, 6, e352.Helms, E.; Onate, M.K.; Sherman, M.H. Fibroblast heterogeneity in the pancreatic tumor microenvironment. Cancer Discov. 2020, 10, 648–656.
- Jacobetz, M.A.; Chan, D.S.; Neesse, A.; Bapiro, T.E.; Cook, N.; Frese, K.K.; Feig, C.; Nakagawa, T.; Caldwell, M.E.; Zecchini, H.I.; et al. Hyaluronan impairs vascular function and drug delivery in a mouse model of pancreatic cancer. Gut 2013, 62, 112–120.Nielsen, M.F.; Mortensen, M.B.; Detlefsen, S. Key players in pancreatic cancer-stroma interaction: Cancer-associated fibroblasts, endothelial and inflammatory cells. World J. Gastroenterol. 2016, 22, 2678–2700.
- Richards, K.E.; Zeleniak, A.E.; Fishel, M.L.; Wu, J.; Littlepage, L.E.; Hill, R. Cancer-associated fibroblast exosomes regulate survival and proliferation of pancreatic cancer cells. Oncogene 2017, 36, 1770–1778.Weniger, M.; Honselmann, K.C.; Liss, A.S. The extracellular matrix and pancreatic cancer: A complex relationship. Cancers 2018, 10, 316.
- Fang, Y.; Zhou, W.; Rong, Y.; Kuang, T.; Xu, X.; Wu, W.; Wang, D.; Lou, W. Exosomal mirna-106b from cancer-associated fibroblast promotes gemcitabine resistance in pancreatic cancer. Exp. Cell Res. 2019, 383, 111543.Mekapogu, A.R.; Pothula, S.P.; Pirola, R.C.; Wilson, J.S.; Apte, M.V. Multifunctional role of pancreatic stellate cells in pancreatic cancer. Ann. Pancreat. Cancer 2019, 2, 10.
- Amrutkar, M.; Vethe, N.T.; Verbeke, C.S.; Aasrum, M.; Finstadsveen, A.V.; Santha, P.; Gladhaug, I.P. Differential gemcitabine sensitivity in primary human pancreatic cancer cells and paired stellate cells is driven by heterogenous drug uptake and processing. Cancers 2020, 12, 3628.Dalin, S.; Sullivan, M.R.; Lau, A.N.; Grauman-Boss, B.; Mueller, H.S.; Kreidl, E.; Fenoglio, S.; Luengo, A.; Lees, J.A.; Vander Heiden, M.G.; et al. Deoxycytidine release from pancreatic stellate cells promotes gemcitabine resistance. Cancer Res. 2019, 79, 5723–5733.
- Masamune, A.; Watanabe, T.; Kikuta, K.; Shimosegawa, T. Roles of pancreatic stellate cells in pancreatic inflammation and fibrosis. Clin. Gastroenterol. Hepatol. 2009, 7, S48–S54.Sherman, M.H.; Yu, R.T.; Tseng, T.W.; Sousa, C.M.; Liu, S.; Truitt, M.L.; He, N.; Ding, N.; Liddle, C.; Atkins, A.R.; et al. Stromal cues regulate the pancreatic cancer epigenome and metabolome. Proc. Natl. Acad. Sci. USA 2017, 114, 1129–1134.
- Zhang, H.; Wu, H.; Guan, J.; Wang, L.; Ren, X.; Shi, X.; Liang, Z.; Liu, T. Paracrine sdf-1alpha signaling mediates the effects of pscs on gem chemoresistance through an il-6 autocrine loop in pancreatic cancer cells. Oncotarget 2015, 6, 3085–3097.Apte, M.V.; Park, S.; Phillips, P.A.; Santucci, N.; Goldstein, D.; Kumar, R.K.; Ramm, G.A.; Buchler, M.; Friess, H.; McCarroll, J.A.; et al. Desmoplastic reaction in pancreatic cancer: Role of pancreatic stellate cells. Pancreas 2004, 29, 179–187.
- Miyamoto, H.; Murakami, T.; Tsuchida, K.; Sugino, H.; Miyake, H.; Tashiro, S. Tumor-stroma interaction of human pancreatic cancer: Acquired resistance to anticancer drugs and proliferation regulation is dependent on extracellular matrix proteins. Pancreas 2004, 28, 38–44.Bachem, M.G.; Schünemann, M.; Ramadani, M.; Siech, M.; Beger, H.; Buck, A.; Zhou, S.; Schmid-Kotsas, A.; Adler, G. Pancreatic carcinoma cells induce fibrosis by stimulating proliferation and matrix synthesis of stellate cells. Gastroenterology 2005, 128, 907–921.
- Shields, M.A.; Dangi-Garimella, S.; Redig, A.J.; Munshi, H.G. Biochemical role of the collagen-rich tumour microenvironment in pancreatic cancer progression. Biochem. J. 2012, 441, 541–552.Hwang, R.F.; Moore, T.; Arumugam, T.; Ramachandran, V.; Amos, K.D.; Rivera, A.; Ji, B.; Evans, D.B.; Logsdon, C.D. Cancer-associated stromal fibroblasts promote pancreatic tumor progression. Cancer Res. 2008, 68, 918–926.
- Januchowski, R.; Swierczewska, M.; Sterzynska, K.; Wojtowicz, K.; Nowicki, M.; Zabel, M. Increased expression of several collagen genes is associated with drug resistance in ovarian cancer cell lines. J. Cancer 2016, 7, 1295–1310.Vonlaufen, A.; Joshi, S.; Qu, C.; Phillips, P.A.; Xu, Z.; Parker, N.R.; Toi, C.S.; Pirola, R.C.; Wilson, J.S.; Goldstein, D.; et al. Pancreatic stellate cells: Partners in crime with pancreatic cancer cells. Cancer Res. 2008, 68, 2085–2093.
- Iseri, O.D.; Kars, M.D.; Arpaci, F.; Gunduz, U. Gene expression analysis of drug-resistant mcf-7 cells: Implications for relation to extracellular matrix proteins. Cancer Chemother. Pharmacol. 2010, 65, 447–455.Coleman, S.J.; Chioni, A.M.; Ghallab, M.; Anderson, R.K.; Lemoine, N.R.; Kocher, H.M.; Grose, R.P. Nuclear translocation of fgfr1 and fgf2 in pancreatic stellate cells facilitates pancreatic cancer cell invasion. EMBO Mol. Med. 2014, 6, 467–481.
- Dangi-Garimella, S.; Krantz, S.B.; Barron, M.R.; Shields, M.A.; Heiferman, M.J.; Grippo, P.J.; Bentrem, D.J.; Munshi, H.G. Three-dimensional collagen i promotes gemcitabine resistance in pancreatic cancer through mt1-mmp-mediated expression of hmga2. Cancer Res. 2011, 71, 1019–1028.Bachem, M.G.; Zhou, S.; Buck, K.; Schneiderhan, W.; Siech, M. Pancreatic stellate cells—role in pancreas cancer. Langenbecks Arch. Surg. 2008, 393, 891–900.
- Dangi-Garimella, S.; Sahai, V.; Ebine, K.; Kumar, K.; Munshi, H.G. Three-dimensional collagen i promotes gemcitabine resistance in vitro in pancreatic cancer cells through hmga2-dependent histone acetyltransferase expression. PLoS ONE 2013, 8, e64566.Schneiderhan, W.; Diaz, F.; Fundel, M.; Zhou, S.; Siech, M.; Hasel, C.; Möller, P.; Gschwend, J.E.; Seufferlein, T.; Gress, T.; et al. Pancreatic stellate cells are an important source of mmp-2 in human pancreatic cancer and accelerate tumor progression in a murine xenograft model and cam assay. J. Cell Sci. 2007, 120, 512–519.
- Liu, Y.; Li, F.; Gao, F.; Xing, L.; Qin, P.; Liang, X.; Zhang, J.; Qiao, X.; Lin, L.; Zhao, Q.; et al. Periostin promotes the chemotherapy resistance to gemcitabine in pancreatic cancer. Tumour. Biol. 2016, 37, 15283–15291.Erkan, M.; Kleeff, J.; Gorbachevski, A.; Reiser, C.; Mitkus, T.; Esposito, I.; Giese, T.; Büchler, M.W.; Giese, N.A.; Friess, H. Periostin creates a tumor-supportive microenvironment in the pancreas by sustaining fibrogenic stellate cell activity. Gastroenterology 2007, 132, 1447–1464.
- Ireland, L.; Santos, A.; Ahmed, M.S.; Rainer, C.; Nielsen, S.R.; Quaranta, V.; Weyer-Czernilofsky, U.; Engle, D.D.; Perez-Mancera, P.A.; Coupland, S.E.; et al. Chemoresistance in pancreatic cancer is driven by stroma-derived insulin-like growth factors. Cancer Res. 2016, 76, 6851–6863.Liu, Y.; Li, F.; Gao, F.; Xing, L.; Qin, P.; Liang, X.; Zhang, J.; Qiao, X.; Lin, L.; Zhao, Q.; et al. Role of microenvironmental periostin in pancreatic cancer progression. Oncotarget 2017, 8, 89552–89565.
- Long, K.B.; Tooker, G.; Tooker, E.; Luque, S.L.; Lee, J.W.; Pan, X.; Beatty, G.L. Il6 receptor blockade enhances chemotherapy efficacy in pancreatic ductal adenocarcinoma. Mol. Cancer Ther. 2017, 16, 1898–1908.Kalluri, R. The biology and function of fibroblasts in cancer. Nat. Rev. Cancer 2016, 16, 582–598.
- Duluc, C.; Moatassim-Billah, S.; Chalabi-Dchar, M.; Perraud, A.; Samain, R.; Breibach, F.; Gayral, M.; Cordelier, P.; Delisle, M.B.; Bousquet-Dubouch, M.P.; et al. Pharmacological targeting of the protein synthesis mtor/4e-bp1 pathway in cancer-associated fibroblasts abrogates pancreatic tumour chemoresistance. EMBO Mol. Med. 2015, 7, 735–753.Tjomsland, V.; Spångeus, A.; Välilä, J.; Sandström, P.; Borch, K.; Druid, H.; Falkmer, S.; Falkmer, U.; Messmer, D.; Larsson, M. Interleukin 1α sustains the expression of inflammatory factors in human pancreatic cancer microenvironment by targeting cancer-associated fibroblasts. Neoplasia 2011, 13, 664–675.
- Neumann, C.C.M.; von Hörschelmann, E.; Reutzel-Selke, A.; Seidel, E.; Sauer, I.M.; Pratschke, J.; Bahra, M.; Schmuck, R.B. Tumor-stromal cross-talk modulating the therapeutic response in pancreatic cancer. Hepatobiliary Pancreat. Dis. Int. 2018, 17, 461–472.Vonlaufen, A.; Phillips, P.A.; Xu, Z.; Goldstein, D.; Pirola, R.C.; Wilson, J.S.; Apte, M.V. Pancreatic stellate cells and pancreatic cancer cells: An unholy alliance. Cancer Res. 2008, 68, 7707–7710.
- Haqq, J.; Howells, L.M.; Garcea, G.; Metcalfe, M.S.; Steward, W.P.; Dennison, A.R. Pancreatic stellate cells and pancreas cancer: Current perspectives and future strategies. Eur. J. Cancer 2014, 50, 2570–2582.Gao, Z.; Wang, X.; Wu, K.; Zhao, Y.; Hu, G. Pancreatic stellate cells increase the invasion of human pancreatic cancer cells through the stromal cell-derived factor-1/cxcr4 axis. Pancreatology 2010, 10, 186–193.
- Müerköster, S.; Wegehenkel, K.; Arlt, A.; Witt, M.; Sipos, B.; Kruse, M.L.; Sebens, T.; Klöppel, G.; Kalthoff, H.; Fölsch, U.R.; et al. Tumor stroma interactions induce chemoresistance in pancreatic ductal carcinoma cells involving increased secretion and paracrine effects of nitric oxide and interleukin-1beta. Cancer Res. 2004, 64, 1331–1337.Tjomsland, V.; Aasrum, M.; Christoffersen, T.; Gladhaug, I.P. Functional heterogeneity in tumor-derived human pancreatic stellate cells: Differential expression of hgf and implications for mitogenic signaling and migration in pancreatic cancer cells. Oncotarget 2017, 8, 71672–71684.
- Armstrong, T.; Packham, G.; Murphy, L.B.; Bateman, A.C.; Conti, J.A.; Fine, D.R.; Johnson, C.D.; Benyon, R.C.; Iredale, J.P. Type i collagen promotes the malignant phenotype of pancreatic ductal adenocarcinoma. Clin. Cancer Res. 2004, 10, 7427–7437.Lenggenhager, D.; Amrutkar, M.; Sántha, P.; Aasrum, M.; Löhr, J.M.; Gladhaug, I.P.; Verbeke, C.S. Commonly used pancreatic stellate cell cultures differ phenotypically and in their interactions with pancreatic cancer cells. Cells 2019, 8, 23.
- Wei, L.; Ye, H.; Li, G.; Lu, Y.; Zhou, Q.; Zheng, S.; Lin, Q.; Liu, Y.; Li, Z.; Chen, R. Cancer-associated fibroblasts promote progression and gemcitabine resistance via the sdf-1/satb-1 pathway in pancreatic cancer. Cell Death Dis. 2018, 9, 1065.Tjomsland, V.; Pomianowska, E.; Aasrum, M.; Sandnes, D.; Verbeke, C.S.; Gladhaug, I.P. Profile of mmp and timp expression in human pancreatic stellate cells: Regulation by il-1alpha and tgfbeta and implications for migration of pancreatic cancer cells. Neoplasia 2016, 18, 447–456.
- Xu, J.; Liu, S.; Yang, X.; Cao, S.; Zhou, Y. Paracrine hgf promotes emt and mediates the effects of psc on chemoresistance by activating c-met/pi3k/akt signaling in pancreatic cancer in vitro. Life Sci. 2020, 263, 118523.Koshiba, T.; Hosotani, R.; Miyamoto, Y.; Ida, J.; Tsuji, S.; Nakajima, S.; Kawaguchi, M.; Kobayashi, H.; Doi, R.; Hori, T.; et al. Expression of stromal cell-derived factor 1 and cxcr4 ligand receptor system in pancreatic cancer: A possible role for tumor progression. Clin. Cancer Res. 2000, 6, 3530–3535.
- Cao, F.; Li, J.; Sun, H.; Liu, S.; Cui, Y.; Li, F. Hes 1 is essential for chemoresistance induced by stellate cells and is associated with poor prognosis in pancreatic cancer. Oncol. Rep. 2015, 33, 1883–1889.Mantoni, T.S.; Schendel, R.R.; Rödel, F.; Niedobitek, G.; Al-Assar, O.; Masamune, A.; Brunner, T.B. Stromal sparc expression and patient survival after chemoradiation for non-resectable pancreatic adenocarcinoma. Cancer Biol. Ther. 2008, 7, 1806–1815.
- Patzak, M.S.; Kari, V.; Patil, S.; Hamdan, F.H.; Goetze, R.G.; Brunner, M.; Gaedcke, J.; Kitz, J.; Jodrell, D.I.; Richards, F.M.; et al. Cytosolic 5′-nucleotidase 1a is overexpressed in pancreatic cancer and mediates gemcitabine resistance by reducing intracellular gemcitabine metabolites. EBioMedicine 2019, 40, 394–405.Marzoq, A.J.; Mustafa, S.A.; Heidrich, L.; Hoheisel, J.D.; Alhamdani, M.S.S. Impact of the secretome of activated pancreatic stellate cells on growth and differentiation of pancreatic tumour cells. Sci. Rep. 2019, 9, 5303.
- Fu, Y.; Liu, S.; Yin, S.; Niu, W.; Xiong, W.; Tan, M.; Li, G.; Zhou, M. The reverse warburg effect is likely to be an achilles’ heel of cancer that can be exploited for cancer therapy. Oncotarget 2017, 8, 57813–57825.Lu, J.; Zhou, S.; Siech, M.; Habisch, H.; Seufferlein, T.; Bachem, M.G. Pancreatic stellate cells promote hapto-migration of cancer cells through collagen i-mediated signalling pathway. Br. J. Cancer 2014, 110, 409–420.
- Kamphorst, J.J.; Nofal, M.; Commisso, C.; Hackett, S.R.; Lu, W.; Grabocka, E.; Vander Heiden, M.G.; Miller, G.; Drebin, J.A.; Bar-Sagi, D.; et al. Human pancreatic cancer tumors are nutrient poor and tumor cells actively scavenge extracellular protein. Cancer Res. 2015, 75, 544–553.Kikuta, K.; Masamune, A.; Watanabe, T.; Ariga, H.; Itoh, H.; Hamada, S.; Satoh, K.; Egawa, S.; Unno, M.; Shimosegawa, T. Pancreatic stellate cells promote epithelial-mesenchymal transition in pancreatic cancer cells. Biochem. Biophys. Res. Commun. 2010, 403, 380–384.
- An, M.X.; Li, S.; Yao, H.B.; Li, C.; Wang, J.M.; Sun, J.; Li, X.Y.; Meng, X.N.; Wang, H.Q. Bag3 directly stabilizes hexokinase 2 mrna and promotes aerobic glycolysis in pancreatic cancer cells. J. Cell Biol. 2017, 216, 4091–4105.Xu, Z.; Vonlaufen, A.; Phillips, P.A.; Fiala-Beer, E.; Zhang, X.; Yang, L.; Biankin, A.V.; Goldstein, D.; Pirola, R.C.; Wilson, J.S.; et al. Role of pancreatic stellate cells in pancreatic cancer metastasis. Am. J. Pathol. 2010, 177, 2585–2596.
- Baek, G.; Tse, Y.F.; Hu, Z.; Cox, D.; Buboltz, N.; McCue, P.; Yeo, C.J.; White, M.A.; DeBerardinis, R.J.; Knudsen, E.S.; et al. Mct4 defines a glycolytic subtype of pancreatic cancer with poor prognosis and unique metabolic dependencies. Cell Rep. 2014, 9, 2233–2249.Liu, S.L.; Cao, S.G.; Li, Y.; Sun, B.; Chen, D.; Wang, D.S.; Zhou, Y.B. Pancreatic stellate cells facilitate pancreatic cancer cell viability and invasion. Oncol. Lett. 2019, 17, 2057–2062.
- Wu, Y.S.; Looi, C.Y.; Subramaniam, K.S.; Masamune, A.; Chung, I. Soluble factors from stellate cells induce pancreatic cancer cell proliferation via nrf2-activated metabolic reprogramming and ros detoxification. Oncotarget 2016, 7, 36719–36732.Swayden, M.; Iovanna, J.; Soubeyran, P. Pancreatic cancer chemo-resistance is driven by tumor phenotype rather than tumor genotype. Heliyon 2018, 4, e01055.
- Pavlides, S.; Vera, I.; Gandara, R.; Sneddon, S.; Pestell, R.G.; Mercier, I.; Martinez-Outschoorn, U.E.; Whitaker-Menezes, D.; Howell, A.; Sotgia, F.; et al. Warburg meets autophagy: Cancer-associated fibroblasts accelerate tumor growth and metastasis via oxidative stress, mitophagy, and aerobic glycolysis. Antioxid. Redox Signal. 2012, 16, 1264–1284.Gnanamony, M.; Gondi, C.S. Chemoresistance in pancreatic cancer: Emerging concepts. Oncol. Lett. 2017, 13, 2507–2513.
- Endo, S.; Nakata, K.; Ohuchida, K.; Takesue, S.; Nakayama, H.; Abe, T.; Koikawa, K.; Okumura, T.; Sada, M.; Horioka, K.; et al. Autophagy is required for activation of pancreatic stellate cells, associated with pancreatic cancer progression and promotes growth of pancreatic tumors in mice. Gastroenterology 2017, 152, 1492–1506.e1424.Adamska, A.; Elaskalani, O.; Emmanouilidi, A.; Kim, M.; Abdol Razak, N.B.; Metharom, P.; Falasca, M. Molecular and cellular mechanisms of chemoresistance in pancreatic cancer. Adv. Biol. Regul. 2018, 68, 77–87.
- Zhao, H.; Yang, L.; Baddour, J.; Achreja, A.; Bernard, V.; Moss, T.; Marini, J.C.; Tudawe, T.; Seviour, E.G.; San Lucas, F.A.; et al. Tumor microenvironment derived exosomes pleiotropically modulate cancer cell metabolism. Elife 2016, 5, e10250.Che, P.P.; Gregori, A.; Firuzi, O.; Dahele, M.; Sminia, P.; Peters, G.J.; Giovannetti, E. Pancreatic cancer resistance conferred by stellate cells: Looking for new preclinical models. Exp. Hematol. Oncol. 2020, 9, 18.
- Ye, L.Y.; Zhang, Q.; Bai, X.L.; Pankaj, P.; Hu, Q.D.; Liang, T.B. Hypoxia-inducible factor 1α expression and its clinical significance in pancreatic cancer: A meta-analysis. Pancreatology 2014, 14, 391–397.Hessmann, E.; Patzak, M.S.; Klein, L.; Chen, N.; Kari, V.; Ramu, I.; Bapiro, T.E.; Frese, K.K.; Gopinathan, A.; Richards, F.M.; et al. Fibroblast drug scavenging increases intratumoural gemcitabine accumulation in murine pancreas cancer. Gut 2018, 67, 497–507.
- Sun, H.C.; Qiu, Z.J.; Liu, J.; Sun, J.; Jiang, T.; Huang, K.J.; Yao, M.; Huang, C. Expression of hypoxia-inducible factor-1 alpha and associated proteins in pancreatic ductal adenocarcinoma and their impact on prognosis. Int. J. Oncol. 2007, 30, 1359–1367.Hesler, R.A.; Huang, J.J.; Starr, M.D.; Treboschi, V.M.; Bernanke, A.G.; Nixon, A.B.; McCall, S.J.; White, R.R.; Blobe, G.C. Tgf-beta-induced stromal cyr61 promotes resistance to gemcitabine in pancreatic ductal adenocarcinoma through downregulation of the nucleoside transporters hent1 and hcnt3. Carcinogenesis 2016, 37, 1041–1051.
- Hoffmann, A.C.; Mori, R.; Vallbohmer, D.; Brabender, J.; Klein, E.; Drebber, U.; Baldus, S.E.; Cooc, J.; Azuma, M.; Metzger, R.; et al. High expression of hif1a is a predictor of clinical outcome in patients with pancreatic ductal adenocarcinomas and correlated to pdgfa, vegf, and bfgf. Neoplasia 2008, 10, 674–679.Provenzano, P.P.; Cuevas, C.; Chang, A.E.; Goel, V.K.; Von Hoff, D.D.; Hingorani, S.R. Enzymatic targeting of the stroma ablates physical barriers to treatment of pancreatic ductal adenocarcinoma. Cancer Cell 2012, 21, 418–429.
- Shibaji, T.; Nagao, M.; Ikeda, N.; Kanehiro, H.; Hisanaga, M.; Ko, S.; Fukumoto, A.; Nakajima, Y. Prognostic significance of hif-1 alpha overexpression in human pancreatic cancer. AntiCancer Res. 2003, 23, 4721–4727.Neesse, A.; Algul, H.; Tuveson, D.A.; Gress, T.M. Stromal biology and therapy in pancreatic cancer: A changing paradigm. Gut 2015, 64, 1476–1484.
- Erkan, M.; Kurtoglu, M.; Kleeff, J. The role of hypoxia in pancreatic cancer: A potential therapeutic target? Expert. Rev. Gastroenterol. Hepatol. 2016, 10, 301–316.Pries, A.R.; Höpfner, M.; le Noble, F.; Dewhirst, M.W.; Secomb, T.W. The shunt problem: Control of functional shunting in normal and tumour vasculature. Nat. Rev. Cancer 2010, 10, 587–593.
- Yuen, A.; Díaz, B. The impact of hypoxia in pancreatic cancer invasion and metastasis. Hypoxia 2014, 2, 91–106.Koay, E.J.; Truty, M.J.; Cristini, V.; Thomas, R.M.; Chen, R.; Chatterjee, D.; Kang, Y.; Bhosale, P.R.; Tamm, E.P.; Crane, C.H.; et al. Transport properties of pancreatic cancer describe gemcitabine delivery and response. J. Clin. Investig. 2014, 124, 1525–1536.
- Rohwer, N.; Cramer, T. Hypoxia-mediated drug resistance: Novel insights on the functional interaction of hifs and cell death pathways. Drug Resist. Updates 2011, 14, 191–201.Stylianopoulos, T.; Martin, J.D.; Chauhan, V.P.; Jain, S.R.; Diop-Frimpong, B.; Bardeesy, N.; Smith, B.L.; Ferrone, C.R.; Hornicek, F.J.; Boucher, Y.; et al. Causes, consequences, and remedies for growth-induced solid stress in murine and human tumors. Proc. Natl. Acad. Sci. USA 2012, 109, 15101–15108.
- Yokoi, K.; Fidler, I.J. Hypoxia increases resistance of human pancreatic cancer cells to apoptosis induced by gemcitabine. Clin. Cancer Res. 2004, 10, 2299–2306.DuFort, C.C.; DelGiorno, K.E.; Carlson, M.A.; Osgood, R.J.; Zhao, C.; Huang, Z.; Thompson, C.B.; Connor, R.J.; Thanos, C.D.; Scott Brockenbrough, J.; et al. Interstitial pressure in pancreatic ductal adenocarcinoma is dominated by a gel-fluid phase. Biophys. J. 2016, 110, 2106–2119.
- Kasuya, K.; Tsuchida, A.; Nagakawa, Y.; Suzuki, M.; Abe, Y.; Itoi, T.; Serizawa, H.; Nagao, T.; Shimazu, M.; Aoki, T. Hypoxia-inducible factor-1alpha expression and gemcitabine chemotherapy for pancreatic cancer. Oncol. Rep. 2011, 26, 1399–1406.Rice, A.J.; Cortes, E.; Lachowski, D.; Cheung, B.C.H.; Karim, S.A.; Morton, J.P.; Del Río Hernández, A. Matrix stiffness induces epithelial-mesenchymal transition and promotes chemoresistance in pancreatic cancer cells. Oncogenesis 2017, 6, e352.
- Yoshida, G.J. Metabolic reprogramming: The emerging concept and associated therapeutic strategies. J. Exp. Clin. Cancer Res. 2015, 34, 111.Jacobetz, M.A.; Chan, D.S.; Neesse, A.; Bapiro, T.E.; Cook, N.; Frese, K.K.; Feig, C.; Nakagawa, T.; Caldwell, M.E.; Zecchini, H.I.; et al. Hyaluronan impairs vascular function and drug delivery in a mouse model of pancreatic cancer. Gut 2013, 62, 112–120.
- Jiang, S.H.; Li, J.; Dong, F.Y.; Yang, J.Y.; Liu, D.J.; Yang, X.M.; Wang, Y.H.; Yang, M.W.; Fu, X.L.; Zhang, X.X.; et al. Increased serotonin signaling contributes to the warburg effect in pancreatic tumor cells under metabolic stress and promotes growth of pancreatic tumors in mice. Gastroenterology 2017, 153, 277–291.e219.Richards, K.E.; Zeleniak, A.E.; Fishel, M.L.; Wu, J.; Littlepage, L.E.; Hill, R. Cancer-associated fibroblast exosomes regulate survival and proliferation of pancreatic cancer cells. Oncogene 2017, 36, 1770–1778.
- Shan, T.; Chen, S.; Chen, X.; Lin, W.R.; Li, W.; Ma, J.; Wu, T.; Cui, X.; Ji, H.; Li, Y.; et al. Cancer-associated fibroblasts enhance pancreatic cancer cell invasion by remodeling the metabolic conversion mechanism. Oncol. Rep. 2017, 37, 1971–1979.Fang, Y.; Zhou, W.; Rong, Y.; Kuang, T.; Xu, X.; Wu, W.; Wang, D.; Lou, W. Exosomal mirna-106b from cancer-associated fibroblast promotes gemcitabine resistance in pancreatic cancer. Exp. Cell Res. 2019, 383, 111543.
- Dunne, R.F.; Hezel, A.F. Genetics and biology of pancreatic ductal adenocarcinoma. Hematol. Oncol. Clin. N. Am. 2015, 29, 595–608.Amrutkar, M.; Vethe, N.T.; Verbeke, C.S.; Aasrum, M.; Finstadsveen, A.V.; Santha, P.; Gladhaug, I.P. Differential gemcitabine sensitivity in primary human pancreatic cancer cells and paired stellate cells is driven by heterogenous drug uptake and processing. Cancers 2020, 12, 3628.
- Tape, C.J.; Ling, S.; Dimitriadi, M.; McMahon, K.M.; Worboys, J.D.; Leong, H.S.; Norrie, I.C.; Miller, C.J.; Poulogiannis, G.; Lauffenburger, D.A.; et al. Oncogenic kras regulates tumor cell signaling via stromal reciprocation. Cell 2016, 165, 1818.Masamune, A.; Watanabe, T.; Kikuta, K.; Shimosegawa, T. Roles of pancreatic stellate cells in pancreatic inflammation and fibrosis. Clin. Gastroenterol. Hepatol. 2009, 7, S48–S54.
- Zhang, H.; Wu, H.; Guan, J.; Wang, L.; Ren, X.; Shi, X.; Liang, Z.; Liu, T. Paracrine sdf-1alpha signaling mediates the effects of pscs on gem chemoresistance through an il-6 autocrine loop in pancreatic cancer cells. Oncotarget 2015, 6, 3085–3097.
- Miyamoto, H.; Murakami, T.; Tsuchida, K.; Sugino, H.; Miyake, H.; Tashiro, S. Tumor-stroma interaction of human pancreatic cancer: Acquired resistance to anticancer drugs and proliferation regulation is dependent on extracellular matrix proteins. Pancreas 2004, 28, 38–44.
- Shields, M.A.; Dangi-Garimella, S.; Redig, A.J.; Munshi, H.G. Biochemical role of the collagen-rich tumour microenvironment in pancreatic cancer progression. Biochem. J. 2012, 441, 541–552.
- Januchowski, R.; Swierczewska, M.; Sterzynska, K.; Wojtowicz, K.; Nowicki, M.; Zabel, M. Increased expression of several collagen genes is associated with drug resistance in ovarian cancer cell lines. J. Cancer 2016, 7, 1295–1310.
- Iseri, O.D.; Kars, M.D.; Arpaci, F.; Gunduz, U. Gene expression analysis of drug-resistant mcf-7 cells: Implications for relation to extracellular matrix proteins. Cancer Chemother. Pharmacol. 2010, 65, 447–455.
- Dangi-Garimella, S.; Krantz, S.B.; Barron, M.R.; Shields, M.A.; Heiferman, M.J.; Grippo, P.J.; Bentrem, D.J.; Munshi, H.G. Three-dimensional collagen i promotes gemcitabine resistance in pancreatic cancer through mt1-mmp-mediated expression of hmga2. Cancer Res. 2011, 71, 1019–1028.
- Dangi-Garimella, S.; Sahai, V.; Ebine, K.; Kumar, K.; Munshi, H.G. Three-dimensional collagen i promotes gemcitabine resistance in vitro in pancreatic cancer cells through hmga2-dependent histone acetyltransferase expression. PLoS ONE 2013, 8, e64566.
- Liu, Y.; Li, F.; Gao, F.; Xing, L.; Qin, P.; Liang, X.; Zhang, J.; Qiao, X.; Lin, L.; Zhao, Q.; et al. Periostin promotes the chemotherapy resistance to gemcitabine in pancreatic cancer. Tumour. Biol. 2016, 37, 15283–15291.
- Ireland, L.; Santos, A.; Ahmed, M.S.; Rainer, C.; Nielsen, S.R.; Quaranta, V.; Weyer-Czernilofsky, U.; Engle, D.D.; Perez-Mancera, P.A.; Coupland, S.E.; et al. Chemoresistance in pancreatic cancer is driven by stroma-derived insulin-like growth factors. Cancer Res. 2016, 76, 6851–6863.
- Long, K.B.; Tooker, G.; Tooker, E.; Luque, S.L.; Lee, J.W.; Pan, X.; Beatty, G.L. Il6 receptor blockade enhances chemotherapy efficacy in pancreatic ductal adenocarcinoma. Mol. Cancer Ther. 2017, 16, 1898–1908.
- Duluc, C.; Moatassim-Billah, S.; Chalabi-Dchar, M.; Perraud, A.; Samain, R.; Breibach, F.; Gayral, M.; Cordelier, P.; Delisle, M.B.; Bousquet-Dubouch, M.P.; et al. Pharmacological targeting of the protein synthesis mtor/4e-bp1 pathway in cancer-associated fibroblasts abrogates pancreatic tumour chemoresistance. EMBO Mol. Med. 2015, 7, 735–753.
- Neumann, C.C.M.; von Hörschelmann, E.; Reutzel-Selke, A.; Seidel, E.; Sauer, I.M.; Pratschke, J.; Bahra, M.; Schmuck, R.B. Tumor-stromal cross-talk modulating the therapeutic response in pancreatic cancer. Hepatobiliary Pancreat. Dis. Int. 2018, 17, 461–472.
- Haqq, J.; Howells, L.M.; Garcea, G.; Metcalfe, M.S.; Steward, W.P.; Dennison, A.R. Pancreatic stellate cells and pancreas cancer: Current perspectives and future strategies. Eur. J. Cancer 2014, 50, 2570–2582.
- Müerköster, S.; Wegehenkel, K.; Arlt, A.; Witt, M.; Sipos, B.; Kruse, M.L.; Sebens, T.; Klöppel, G.; Kalthoff, H.; Fölsch, U.R.; et al. Tumor stroma interactions induce chemoresistance in pancreatic ductal carcinoma cells involving increased secretion and paracrine effects of nitric oxide and interleukin-1beta. Cancer Res. 2004, 64, 1331–1337.
- Armstrong, T.; Packham, G.; Murphy, L.B.; Bateman, A.C.; Conti, J.A.; Fine, D.R.; Johnson, C.D.; Benyon, R.C.; Iredale, J.P. Type i collagen promotes the malignant phenotype of pancreatic ductal adenocarcinoma. Clin. Cancer Res. 2004, 10, 7427–7437.
- Wei, L.; Ye, H.; Li, G.; Lu, Y.; Zhou, Q.; Zheng, S.; Lin, Q.; Liu, Y.; Li, Z.; Chen, R. Cancer-associated fibroblasts promote progression and gemcitabine resistance via the sdf-1/satb-1 pathway in pancreatic cancer. Cell Death Dis. 2018, 9, 1065.
- Xu, J.; Liu, S.; Yang, X.; Cao, S.; Zhou, Y. Paracrine hgf promotes emt and mediates the effects of psc on chemoresistance by activating c-met/pi3k/akt signaling in pancreatic cancer in vitro. Life Sci. 2020, 263, 118523.
- Cao, F.; Li, J.; Sun, H.; Liu, S.; Cui, Y.; Li, F. Hes 1 is essential for chemoresistance induced by stellate cells and is associated with poor prognosis in pancreatic cancer. Oncol. Rep. 2015, 33, 1883–1889.
- Patzak, M.S.; Kari, V.; Patil, S.; Hamdan, F.H.; Goetze, R.G.; Brunner, M.; Gaedcke, J.; Kitz, J.; Jodrell, D.I.; Richards, F.M.; et al. Cytosolic 5′-nucleotidase 1a is overexpressed in pancreatic cancer and mediates gemcitabine resistance by reducing intracellular gemcitabine metabolites. EBioMedicine 2019, 40, 394–405.
- An, M.X.; Li, S.; Yao, H.B.; Li, C.; Wang, J.M.; Sun, J.; Li, X.Y.; Meng, X.N.; Wang, H.Q. Bag3 directly stabilizes hexokinase 2 mrna and promotes aerobic glycolysis in pancreatic cancer cells. J. Cell Biol. 2017, 216, 4091–4105.
- Baek, G.; Tse, Y.F.; Hu, Z.; Cox, D.; Buboltz, N.; McCue, P.; Yeo, C.J.; White, M.A.; DeBerardinis, R.J.; Knudsen, E.S.; et al. Mct4 defines a glycolytic subtype of pancreatic cancer with poor prognosis and unique metabolic dependencies. Cell Rep. 2014, 9, 2233–2249.
- Wu, Y.S.; Looi, C.Y.; Subramaniam, K.S.; Masamune, A.; Chung, I. Soluble factors from stellate cells induce pancreatic cancer cell proliferation via nrf2-activated metabolic reprogramming and ros detoxification. Oncotarget 2016, 7, 36719–36732.
- Pavlides, S.; Vera, I.; Gandara, R.; Sneddon, S.; Pestell, R.G.; Mercier, I.; Martinez-Outschoorn, U.E.; Whitaker-Menezes, D.; Howell, A.; Sotgia, F.; et al. Warburg meets autophagy: Cancer-associated fibroblasts accelerate tumor growth and metastasis via oxidative stress, mitophagy, and aerobic glycolysis. Antioxid. Redox Signal. 2012, 16, 1264–1284.
- Endo, S.; Nakata, K.; Ohuchida, K.; Takesue, S.; Nakayama, H.; Abe, T.; Koikawa, K.; Okumura, T.; Sada, M.; Horioka, K.; et al. Autophagy is required for activation of pancreatic stellate cells, associated with pancreatic cancer progression and promotes growth of pancreatic tumors in mice. Gastroenterology 2017, 152, 1492–1506.e1424.
- Zhao, H.; Yang, L.; Baddour, J.; Achreja, A.; Bernard, V.; Moss, T.; Marini, J.C.; Tudawe, T.; Seviour, E.G.; San Lucas, F.A.; et al. Tumor microenvironment derived exosomes pleiotropically modulate cancer cell metabolism. Elife 2016, 5, e10250.
- Ye, L.Y.; Zhang, Q.; Bai, X.L.; Pankaj, P.; Hu, Q.D.; Liang, T.B. Hypoxia-inducible factor 1α expression and its clinical significance in pancreatic cancer: A meta-analysis. Pancreatology 2014, 14, 391–397.
- Sun, H.C.; Qiu, Z.J.; Liu, J.; Sun, J.; Jiang, T.; Huang, K.J.; Yao, M.; Huang, C. Expression of hypoxia-inducible factor-1 alpha and associated proteins in pancreatic ductal adenocarcinoma and their impact on prognosis. Int. J. Oncol. 2007, 30, 1359–1367.
- Hoffmann, A.C.; Mori, R.; Vallbohmer, D.; Brabender, J.; Klein, E.; Drebber, U.; Baldus, S.E.; Cooc, J.; Azuma, M.; Metzger, R.; et al. High expression of hif1a is a predictor of clinical outcome in patients with pancreatic ductal adenocarcinomas and correlated to pdgfa, vegf, and bfgf. Neoplasia 2008, 10, 674–679.
- Shibaji, T.; Nagao, M.; Ikeda, N.; Kanehiro, H.; Hisanaga, M.; Ko, S.; Fukumoto, A.; Nakajima, Y. Prognostic significance of hif-1 alpha overexpression in human pancreatic cancer. AntiCancer Res. 2003, 23, 4721–4727.
- Erkan, M.; Kurtoglu, M.; Kleeff, J. The role of hypoxia in pancreatic cancer: A potential therapeutic target? Expert. Rev. Gastroenterol. Hepatol. 2016, 10, 301–316.
- Yuen, A.; Díaz, B. The impact of hypoxia in pancreatic cancer invasion and metastasis. Hypoxia 2014, 2, 91–106.
- Rohwer, N.; Cramer, T. Hypoxia-mediated drug resistance: Novel insights on the functional interaction of hifs and cell death pathways. Drug Resist. Updates 2011, 14, 191–201.
- Yokoi, K.; Fidler, I.J. Hypoxia increases resistance of human pancreatic cancer cells to apoptosis induced by gemcitabine. Clin. Cancer Res. 2004, 10, 2299–2306.
- Kasuya, K.; Tsuchida, A.; Nagakawa, Y.; Suzuki, M.; Abe, Y.; Itoi, T.; Serizawa, H.; Nagao, T.; Shimazu, M.; Aoki, T. Hypoxia-inducible factor-1alpha expression and gemcitabine chemotherapy for pancreatic cancer. Oncol. Rep. 2011, 26, 1399–1406.
- Yoshida, G.J. Metabolic reprogramming: The emerging concept and associated therapeutic strategies. J. Exp. Clin. Cancer Res. 2015, 34, 111.
- Jiang, S.H.; Li, J.; Dong, F.Y.; Yang, J.Y.; Liu, D.J.; Yang, X.M.; Wang, Y.H.; Yang, M.W.; Fu, X.L.; Zhang, X.X.; et al. Increased serotonin signaling contributes to the warburg effect in pancreatic tumor cells under metabolic stress and promotes growth of pancreatic tumors in mice. Gastroenterology 2017, 153, 277–291.e219.
- Shan, T.; Chen, S.; Chen, X.; Lin, W.R.; Li, W.; Ma, J.; Wu, T.; Cui, X.; Ji, H.; Li, Y.; et al. Cancer-associated fibroblasts enhance pancreatic cancer cell invasion by remodeling the metabolic conversion mechanism. Oncol. Rep. 2017, 37, 1971–1979.
- Dunne, R.F.; Hezel, A.F. Genetics and biology of pancreatic ductal adenocarcinoma. Hematol. Oncol. Clin. N. Am. 2015, 29, 595–608.
- Tape, C.J.; Ling, S.; Dimitriadi, M.; McMahon, K.M.; Worboys, J.D.; Leong, H.S.; Norrie, I.C.; Miller, C.J.; Poulogiannis, G.; Lauffenburger, D.A.; et al. Oncogenic kras regulates tumor cell signaling via stromal reciprocation. Cell 2016, 165, 1818.