Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is a great threat to public health, being a causative pathogen of a deadly coronavirus disease (COVID-19). It has spread to more than 200 countries and infected millions of individuals globally. Although SARS-CoV-2 has structural/genomic similarities with the previously reported SARS-CoV and MERS-CoV, the specific mutations in its genome make it a novel virus. Available therapeutic strategies failed to control this virus. Despite strict standard operating procedures (SOPs), SARS-CoV-2 has spread globally and it is mutating gradually as well.
- SARS-CoV-2
- pandemic
- genomic characterization
- pathophysiology
- therapeutic strategies
- COVID-19 vaccines
Note: The following contents are extract from your paper. The entry will be online only after author check and submit it.
1. Introduction
The emergence and re-emergence of pathogens is a global human health concern [1]. Coronaviruses are enveloped, their genomes are non-segmented, and they are single-stranded positive-sense RNA (+ssRNA) viruses belonging to the family Coronaviridae and order Nidovirales, which are widely dispersed in humans, animals, and birds. Coronaviruses cause various life-threatening diseases from respiratory infections to hepatic, enteric, and severe neurological diseases [2][3][2,3]. Six species of Coronaviruses were known to cause human diseases [4], out of which four (HKU1, NL63, 229E, and OC43) are widespread and responsible for the common cold in individuals with a weak immune response [4]. SARS-CoV-2 is the seventh coronavirus known to infect humans. Its exact origin is unknown; however, it shows homology with the previously identified coronavirus strains SARS-CoV (intermediate host, masked palm civet) and MERS-CoV (intermediate host, dromedary camel) [5][6][5,6]. The homology between SARS-CoV-2 and SARS-CoV is 82.45%, and the homology between SARS-CoV-2 and MERS-CoV is 69.58% [7]. SARS-CoV was responsible for SARS outbreaks in 2002–03 in Guangdong Province, China [8][9][10][8–10], while MERS-CoV was responsible for respiratory illness in the Middle East in 2012–13 [11]. The mortality rates of MERS and SARS were 37% and 10%, respectively [12][13][12,13]. SARS-CoV-2 triggered the COVID-19 pandemic, which spread rapidly worldwide and has become a public health concern [14] [14].
2. Insights into Genomic Organization
Coronaviruses, which belong to the Coronaviridae family, are enveloped and pleomorphic viruses [15]. These are positive-sense RNA viruses with a genome size of 30 kb; which appears to be the largest size for a RNA virus, containing a 5′ cap and 3′ poly A-tail. Coronaviruses have a helical and flexible nucleocapsid. The membrane of these viruses contains a membrane glycoprotein, enveloped protein, and spike protein while the RNA is surrounded by nucleocapsid [16][17][16,17].
Virus RNA contains 6 open reading frames (ORF1ab, ORF3a, ORF6, ORF7ab, ORF8, and ORF10). Two-thirds of the virus genome comprises 1a/1b ORF and the remaining one-third of the genome code is used for M (membrane), S (spike), N (nucleocapsid), and E (enveloped) viral structural proteins [18][19][18,19].
Transcription was carried out by the synthesis of sgRNA (sub-genomic RNA) and replication-transcription complex (RTC), enveloped in double-membrane vesicles. Transcription termination occurred through transcription regulatory sequences that are present in between open reading frames (ORFs). There are 6 ORFs in the SARS-CoV-2 genome, as discussed above [18]. A frameshift mutation in ORF1a and ORF1b produces polypeptides (pp1a and pp1ab), which are further processed by virally encoded proteases such as main proteases (Mpro), chymotrypsin-like proteases (3CLpro), or by papain-like proteases for the production of non-structural proteins (nsps) [20][21][20,21]. Besides 1a and 1b open reading frames (ORFs), all other ORFs are responsible for the production of structural proteins (membrane, nucleocapsid, enveloped, and spike proteins), as shown in Figure 1.
Through sequence analysis of SARS-CoV-2 and SARS-CoV, scientists proposed a mutation in the spike protein responsible for the jumping of the virus from animals to humans [22]. Similarly, some mutations have also been found in protein sequences which lead to the formation of proteins with a change in amino acid residues. For example, at position 723, instead of glycine there is a serine, while at position 1010 there is proline instead of isoleucine [22]. Potential disease recurrence depends on the evolution of the virus due to the accumulation of mutations in the viral genome over time.
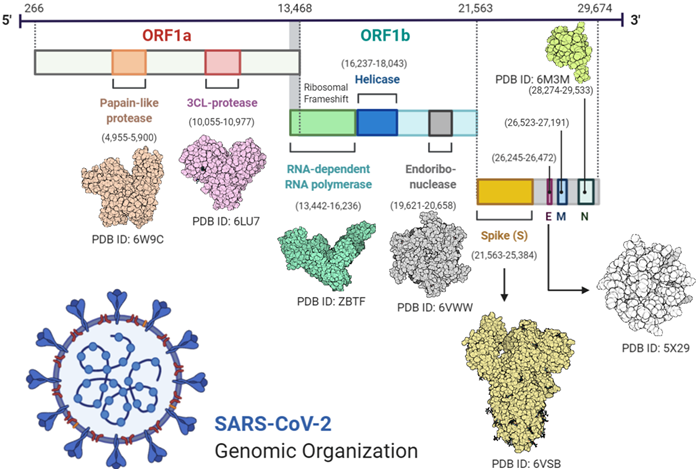
Figure 1. Complete structural and genomic organization of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) [23].
2.1. Genome Sequencing
Through genomic sequence analysis, it has been confirmed that although SARS-CoV-2 has many similarities with SARS-CoV and other related coronaviruses, it is a novel virus (Table 1). The virus made a shift in the host organism from animals to humans with a few unique modifications/mutations. Genome sequence analysis suggests that most of the viral contigs/reads had a similarity with the genome of beta-coronavirus. SARS-CoV-2 has 96.20% and 88.00% levels of similarity to the previously published SARSr-CoV (RaTG13) and bat-SL-CoVZC45 genomes, respectively [3]. The sequencing of the SARS-CoV-2 genome from another study indicated 69.58% and 82.45% sequence similarity with MERS-CoV and SARS-CoV genomes, respectively [24][5,24]. Ten viral genome sequences obtained from 9 patients exhibited 99.98% sequence identity. In another study, sequences from eight patient samples had 99.98% sequence identity with each other across the whole genome [24]. BLASTn search of SARS-CoV-2 sequences has identified matches from the most closely related previously known viruses: SARS-like beta-coronavirus of bat origin, bat-SL-CoVZC45 (sequence identity 88%; query coverage 99%), and bat-SL-CoVZXC21 (sequence identity 88%; query coverage 98%). In 5 gene regions (7, M, N, 14, and E), sequence identity was more than 90% with 98.7% as the highest level for the envelope (E) gene. The Spike (S) gene demonstrated the lowest sequence identity of 75%. However, the sequence identity in 1a and 1b gene regions was 90% and 87%, respectively [24]. The majority of proteins encoded by SARS-CoV-2 were highly similar to proteins encoded by bat-related coronaviruses with a few insertions and deletions [24]. However, protein 13 and the S protein revealed 73.2% and 80% identity with other bat-derived viral proteins, respectively [25]. SARS-CoV-2 encoded a large spike protein, which is a major distinguishing feature among SARS-CoV-2, SARS-CoV, MERS-CoV, and other bat-derived coronaviruses. SARS-CoV-2 exhibits the same genomic organization as bat-SL-CoVZXC21, SARS-CoV, and bat-SL-CoVZC45, as revealed by comparison of predicted coding regions. Ten coding regions were identified including E, M, N, S, 10ab, 9, 8, 7, 3, and 1ab [24].
Table 1. Sequence homology between SARS-CoV-2 and other coronaviruses strains [7].
|
Coronaviruses Strains |
Sequence Similarity |
|
SARSr-CoV; RaTG13 |
96.20% |
|
bat-SL-CoVZC45 |
88.00% |
|
bat-SL-CoVZXC21 |
88.00% |
|
SARS-CoV |
82.45% |
|
SARS-HCoV Tor2 |
82.00% |
|
SARS-HCoV BJ01 |
82.00% |
|
MERS-CoV |
69.58% |
|
HCoV-OC43 |
68.93% |
|
HCoV-HKU1 |
67.59% |
|
HCoV-229E |
65.04% |
|
HCoV-NL63 |
65.11% |
2.2. Phylogenetic Analysis
Phylogenetic analysis of SARS-CoV-2 genomes obtained from early patient samples suggested similarity in the sequence organization with beta-coronaviruses such as 5′ UTR (untranslated region), replicase complex (orf1ab), 4 genes (M, N, S, and E), 3′ UTRs (untranslated regions1), and some unidentified non-structural ORFs (open reading frames) [26]. Instead of having sequence similarity with beta-coronaviruses discovered in bats, SARS-CoV-2 is distinct from SARS-CoV, as well as MERS-CoV. Another piece of evidence pointing to its novelty is that the sequence identity in conserved replicase domains (ORF 1ab) is less than 90% between SARS-CoV-2 and other members of beta-coronaviruses and sarbeco-virus sub-genus of the Coronaviridae family [3].
2.3. Conserved Proteins
The S protein is responsible for membrane fusion and receptor binding. It is also critical in controlling virus transmission capacity and host tropism. The S protein of SARS-CoV-2 has two domains, namely the S1 and S2 domains. The S1 domain is responsible for receptor binding, while the S2 domain for membrane fusion [27]. It has been reported that a cellular protease (furin) is responsible for the cleavage of S1/S2 sites and this cleavage is necessary for the entry of virion in human lung cells and S protein facilitated cell fusion [28]. The S1 and S2 domains of SARS-CoV-2 have a sequence similarity of 93% and 68% with bat-SL-CoVZXC21 and bat-SL-CoVZC45, respectively [29][24,29]. Among sarbeco-coronaviruses, amino acid variations in S protein were identified. Although SARS-CoV and SARS-CoV-2 belong to different clades in the phylogenetic tree, they have 50 conserved amino acids in the S1 domain of the S protein. However, MERS-CoV has mutational differences in S proteins. Most of these mutational events occur in the C-terminal domain [24]. Several other proteases are also involved in different processes, such as entry of the virion, maturation of polyprotein, and assembly of different virion particles [30]. Other than the S protein, a variety of SARS-CoV-2 other proteins show similarity with proteins of other Coronaviridae family members, as shown in Table 2.
Table 2. Percentage identity between proteins of SARS-CoV-2 and the Coronaviridae family [31].
|
Gene |
SARS NC_004718.3 |
Bat MG772934.1 |
Bat DQ022305.2 |
|
ORF1ab |
86.12% |
95.15% |
85.78% |
|
ORF3a |
72.36% |
92.00% |
72.99% |
|
ORF6 |
68.85% |
93.44% |
67.21% |
|
ORF7a |
85.25% |
88.43% |
88.52% |
|
ORF7b |
81.40% |
93.02% |
79.07% |
|
ORF8 |
30.16% |
94.21% |
57.02% |
|
ORF10 |
72.45% |
73.20% |
74.23% |
|
S (Spike) |
75.96% |
80.32% |
76.04% |
|
E (Envelope) |
94.74% |
100% |
94.74% |
|
M (Membrane) |
90.54% |
98.65% |
90.99% |
|
N (Nucleo-capsid) |
90.52% |
94.27% |
89.55% |
2.4. Receptor Binding Domain (RBD)
The RBD of SARS-CoV-2 is found in the C-terminal domain of spike protein as in SARS-CoV, Bat CoV HKU4, and MERS-CoV [32][33] [32,33]. It was also reported that SARS-CoV-2 uses ACE2 (angiotensin-converting enzyme) as a cell receptor for entry into the human cells [34]. From the phylogenetic analysis, it was found that at genome level, SARS-CoV-2 is closely related to bat-SL-CoVZXC21 and bat-SL-CoVZC45, though the RBD of SARS-CoV-2 is highly similar to SARS-CoV. However, key residues of the receptor-binding domain responsible for the binding of the receptor were different in SARS-CoV-2 as compared to SARS-CoV [24]. From the above studies, it is again established that although SARS-CoV-2 has a great similarity with MERS-CoV, SARS-CoV, and some other bat-derived coronaviruses, it is a novel version of coronavirus and is responsible for an infection that is spreading globally.