UA is an independent predictor of mortality in acute and chronic HF, making it a significant prognostic factor in both settings. High serum levels have been also associated with an increased incidence of HF, thus expanding the clinical utility of UA.
- uric acid
- heart failure
- biomarkers
1. Introduction
With a worse prognosis than breast cancer in women and bladder cancer in men, heart failure (HF) represents a major global health problem [1][2][1,2]. This complex clinical entity, commonly defined as the inability of a heart to fulfil required metabolic demands and the perfusion of organs and tissues due to structural or functional cardiac abnormalities, is actually the most common cause of hospitalization after normal delivery, affecting more than 26 million people worldwide [3][4][3,4]. Major population studies reported stable incidence but an increase in HF prevalence and only a slight decrease in HF-related mortality in various populations, with a rather intriguing twist: a trend of a slight rise of HF-related mortality recently [5][6][7][8][5,6,7,8]. The observed increase in prevalence with stable incidence could be explained by the ageing population and improvements in HF treatment [2]. However, this rise will inevitably cause further increases in hospitalization rates and consequently, health care expenditures. Based on available data, experts agree that there is an urgent need for a cost-effective prognostic biomarker in HF. A significant number of biomarkers have already been investigated in the setting of HF [9].
2. Underlying Molecular Mechanisms of HF Development
Virtually any disease or defect that impairs heart structure or function can subsequently lead to HF development. Although most commonly caused by coronary artery disease (CAD), unregulated diabetes, and hypertension, a palette of other etiologic factors, both intra- and extracardiac, can induce HF development [10][15].
In multiple pathophysiological pathways that are operative in HF, such as myocardial necrosis, upregulation of the renin-angiotensin-aldosterone system (RAAS), overt activation of the sympathetic nervous system, and endothelial dysfunction, a recently recognized pathologic process of endothelial-to-mesenchymal transition (EndoMT) emerged as a potent pathobiological driver of pro-fibrotic signaling pathways in HF, thus leading to myocardial fibrosis and adverse ventricular remodeling [11][12][13][14][15][16][17][18][16,17,18,19,20,21,22,23]. EndoMT is a dynamic shift in endothelial cell phenotype toward mesenchymal cells such as myofibroblasts, smooth muscle cells, and osteoblasts [19][24]. EndoMT-mediated fibrosis seems to be driven mainly by TGF-β via SMAD-2/3/4 and the Slug signaling pathway [20][21][25,26]. Hence, it has been hypothesized that EndoMT represents an integrative pathophysiological crosstalk between inflammation and fibrosis, making it a potential therapeutic target in HF [22][27]. Apart from being implicated in cardiac fibrosis, recent evidence indicates that EndoMT plays a role in several cardiac pathologies, including pulmonary artery hypertension, atherosclerosis, endocardial fibroelastosis, and valvular heart disease [23][24][25][26][27][28,29,30,31,32].
In 2013, Paulus and Tschöpe elaborated a pathophysiological model of HF with preserved ejection fraction (HFpEF), which proposes that highly prevalent co-morbidities of HF such as ageing, diabetes mellitus, metabolic syndrome, salt-sensitive hypertension, atrial fibrillation (AF), anemia, chronic obstructive pulmonary disease, and especially obesity exert their detrimental effects on the heart via endothelium of coronary microcirculation, which, as hypothesized, acts as a sort of central processing unit and transfers damage to the heart [28][33]. According to this model, the mentioned comorbidities induce a systemic pro-inflammatory state and thereby stimulate endothelial cells on reactive oxygen species (ROS) production [29][34]. Consequently, ROS trigger cardiomyocyte autophagy, apoptosis, or necrosis and reduce nitric oxide (NO) bioavailability, leading to endothelial dysfunction [28][33]. Importantly, ROS-mediated impaired nitric oxide-cyclic guanosine monophosphate-protein kinase G (NO-cGMP-PKG) signaling also leads to a rise in the resting tension of cardiomyocytes (i.e., myocardial stiffness) via hypophosphorylation of titin [30][31][32][33][35,36,37,38].
3. Molecular Mechanisms by Which UA Is Implicated in Pathophysiology of HF
UA is the end product of both dietary and endogenous purine metabolism in humans [34][39]. Due to the loss of uricase, an enzyme responsible for UA conversion into allantoin, humans are exposed to >50 times greater serum uric acid (SUA) concentrations than other mammals, making them susceptible to hyperuricemic repercussions [35][40]. Although from an evolutionary point of view the loss of uricase may have provided a survival advantage by amplifying the effects of fructose to enhance fat stores and by increasing blood pressure in response to salt, the absence of uricase gene expression, often referred to as “thrifty,” may have exhibited a range of detrimental effects on modern humans owing to the change in diet [36][41]. As a matter of fact, Neel et al. hypothesized that the loss of uricase could at least in part explain the current epidemic of obesity and diabetes [37][42]. Another evolutionary advantage of UA was proposed by Ames et al., who demonstrated that UA is a powerful scavenger of free radicals [38][43]. Figures suggest that UA contributes as much as 60% of free radical scavenging in human serum [39][44]. Moreover, systemic administration of UA increases plasma antioxidant capacity both at rest and after exercise in healthy volunteers [40][41][45,46].
Nevertheless, the biological effects of UA regarding oxidative stress are rather confounding. Unlike the antioxidant effects that UA exerts in extracellular, hydrophilic milieu, intracellularly it imposes detrimental effects, acting as a pro-oxidant [42][47]. Multiple experimental studies demonstrated that UA stimulated ROS creation in various cells, including endothelial cells, vascular smooth muscle cells (VSMCs), hepatocytes, and renal tubular cells, each with a set of repercussions. In endothelial cells it results in decreased NO bioavailability and inhibited cell migration and proliferation, whereas in hepatocytes it results in intracellular fat accumulation [43][44][48,49]. Furthermore, UA activates pro-inflammatory pathways and stimulates cell proliferation in VSMCs, stimulates EndoMT in renal tubular cells, and supports insulin resistance by generating oxidative stress in adipocytes [45][46][47][50,51,52]. Findings that implicate the pro-oxidative activity of UA are further substantiated by the protective effects of probenecid, an inhibitor of the organic anion transporter, which blocks the entry of UA into the cells and ameliorates oxidative stress [46][51]. Kang et al. tried to elucidate this peculiar dual role of UA in oxidative stress by the presence of an unrecognized molecular switch that controls the role of UA acting as a pro-oxidant or as an anti-oxidant [42][47]. Regarding the direct effects of UA on cardiomyocytes, multiple studies demonstrated that hyperuricemia inhibits myocardial cell activity by activating the extracellular signal-regulated kinase (ERK)/P38 signaling pathway through oxidative stress in vitro and induces cardiomyocyte apoptosis through the activation of calpain-1 and endoplasmic reticulum stress in rats [47][48][49][50][53,54,55]. Conversely, a study in healthy men showed that acute exposure to high levels of UA had no effect on hemodynamic variables, basal forearm blood flow, or nitric oxide-dependent endothelial function, implying that UA does not impair cardiovascular function [35][40]. Taken together, the direct effects of UA on the heart still remain quite ambiguous.
The last two steps of purine metabolism are catalyzed with xanthine oxidase (XO) [51][56]. The mentioned organic chemical reactions catalyzed by XO also generate free radicals as a byproduct [52][57]. Interestingly, XO was actually the first identified biological system to produce ROS and is in fact one of the strongest known sources of ROS production in human physiology [53][58]. In physiological milieu, XO-derived reactive oxygen species may have favorable effects, such as modulation of systemic redox balance and a line of defense against bacterial infections [54][59]. Conversely, overexpression of XO could have ROS-mediated detrimental effects such as endothelial function, inflammatory activation, mitochondrial damage, or impaired cardiac contractility, all of which are commonly seen in HF. Since the involvement of ROS in the development of HF has been well documented, multiple authors investigated the role of XO in HF pathophysiology in both animal and human studies [55][60]. XO upregulation in HF could be explained by commonly observed events in HF, such as hypoxia, increased catabolism, cell death, and insulin resistance, which lead to purine degradation and a subsequent increase in substrate supply [56][57][61,62]. In line with this, as demonstrated by multiple authors, a direct assessment of enzyme activity showed that XO activity was extremely upregulated (up to tenfold) in HF [58][59][60][63,64,65]. Studies suggest that XO is involved in HF development via endothelial dysfunction, myocyte apoptosis, and cardiac mechano-energetic coupling. Endothelial dysfunction is a direct result of an increase in the production of ROS as a consequence of XO upregulation in HF, as we noted in the previous section [61][66]. Additional evidence to support this notion is that the administration of allopurinol, a well-known XO inhibitor, improves endothelial dysfunction while reducing markers of oxidative stress among patients with HF [62][67]. In addition, Leyver et al. reported an inverse relationship between SUA and VO2 max and a positive correlation between UA levels and minute ventilation/carbon dioxide production (VE/VCO2), both of which suggest that increased SUA concentrations may reflect an impairment of the oxidative metabolism with consequent exercise intolerance in HF [63][68]. Apart from the abovementioned deleterious effects, XO upregulation is associated with increased filling pressures in systolic HF, diastolic dysfunction, and cachexia [64][65][66][69,70,71]. XO also emerged as a critical factor in upregulating myocardial apoptosis, a central feature in the progression of HF [67][72]. Finally, studies suggest that XO impedes the mechano-energetic uncoupling of the heart via crosstalk with cardiac NO signaling pathways. Mechano-energetic coupling is a phenomenon in a failing heart that implies that despite significantly impaired left ventricular LV work, the oxygen consumed for myocardial contraction remains relatively unchanged, resulting in a decrease in the mechanical efficiency of contractions [68][73]. It has been demonstrated that the administration of allopurinol in dogs with HF decreases oxygen consumption and increases myocardial contractility at both rest and exercise, as well as in response to the stimulation of dobutamine, all of the effects being limited to a failing heart [69][70][74,75]. In concordance with animal studies, Cappola et al. demonstrated that allopurinol administration can improve myocardial efficiency by decreasing oxygen consumption without simultaneous impairment in cardiac function [71][76]. Interestingly, these effects were abrogated by being blocked by N(G)-monomethyl L-arginine (L-NMMA), an NO synthase inhibitor, implicating the importance of NO signaling pathways in this process [70][75].
In the HF setting, elevated SUA levels are owed to at least two distinct mechanisms (Figure 1). The first is increased production and the latter is reduced excretion of UA. The former is caused by both a substantial increase in XO activity and increased oxidative stress, which arises from reduced tissue perfusion and altered metabolic state [51][67][56,72]. Conversely, multiple mechanisms lead to reduced kidney UA excretion. Functional renal impairment, as a part of cardiorenal syndrome, leads to decreased UA excretion in the kidney [72][73][77,78]. Furthermore, diuretics, which are widely prescribed to HF patients, lead to substantial loss of water and salt, thus stimulating proximal tubule reabsorption and a subsequent rise in SUA [74][79]. Other medications, such as noradrenaline and angiotensin II, can also promote hyperuricemia by stimulating UA tubular absorption [75][80]. In a state of impaired muscle perfusion and a consequent switch to an anaerobic metabolism such as HF, lactic acid plasma levels increase and lead to hyperuricemia by further mitigating UA renal excretion [76] [81]. Ultimately, elevated UA itself can impair renal function, creating a positive feedback loop [77][82]. Of important note, patients with ischemic and non-ischemic HF show a similar distribution of SUA concentrations with respect to New York Heart Association (NYHA) classes [78][83]. This highlights the significant role of UA in HF independent of the presence of the metabolic syndrome, a common risk profile for ischemic heart disease, thus bringing further evidence that supports the notion that hyperuricemia is an intrinsic feature within the HF pathophysiology. Since SUA levels correlate with poor clinical outcomes of chronic HF (CHF) in a more evident manner among patients with impaired renal function, it seems that increased UA synthesis is a more significant contributor to the observed elevation of SUA in CHF than a reduction in UA excretion [79][84]. Conversely, Park et al. argued that cell death caused by ischemic insult or acute deterioration of renal function in acute heart failure (AHF) may be the dominant factor for hyperuricemia in that setting [80][85].
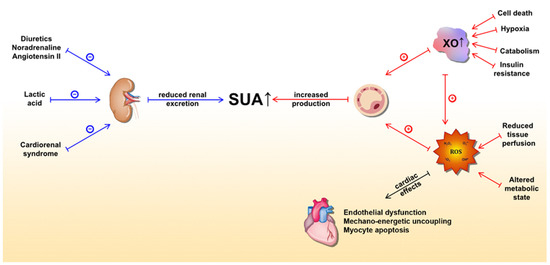
Figure 1. Underlying molecular mechanisms of serum uric acid (SUA) elevation in heart failure and detrimental effects of xanthine oxidase activity (mediated by ROS) on the heart. The blue lines represent mechanisms that lead to a reduction in UA excretion, whereas the red lines represent mechanisms leading to increased UA production. Abbreviations: SUA: serum uric acid; XO: xanthine oxidase; ROS: reactive oxygen species.