Viruses require a host for replication and survival and hence are subjected to host immunological pressures. The complement system, a crucial first response of the host immune system, is effective in targeting viruses and virus-infected cells, and boosting the antiviral innate and acquired immune responses. Thus, the system imposes a strong selection pressure on viruses. Consequently, viruses have evolved multiple countermeasures against host complement. A major mechanism employed by viruses to subvert the complement system is encoding proteins that target complement. Since viruses have limited genome size, most of these proteins are multifunctional in nature.
- viral immune evasion
- complement
- innate immunity
- pathogenesis
- Poxvirus
- Herpesvirus
- RCA
- CD59
- viral RCA
Note: The following contents are extract from your paper. The entry will be online only after author check and submit it.
1. Introduction
Viruses face constant challenges from the host due to host resistance, and the environment because of variations in temperature and humidity. Nonetheless, they adapt to these dynamic changes to become the most successful pathogens [1]. The success of virus infection and propagation is determined by many host factors as well as host-virus interactions.
The concept of host resistance to infection was first documented by the Greek historian Thucydides, who provided an eye-witness account of the plague that struck Athens between 430–427 B.C. [2]. However, it was only in the early 20th century that Mechnikov developed the theory of cellular immunity and Ehrlich established the idea of humoral immunity. Yet another critical observation that was made by multiple researchers during the late 19th century was a bactericidal activity of normal serum. Following these studies, in 1895, Bordet showed that bactericidal activity is owing to antibody and a preexisting ‘heat-labile component’ (later dubbed as complement by Ehrlich), which marked the beginning of complement research [3]. He also developed the complement fixation test [4], which became a valuable tool for the diagnosis of various viral diseases and establishing antigenic relationship among viruses. In 1919, he received the Nobel Prize in Medicine for his pioneering work.
The 20th century saw the beginning of the rise of the complement field. It became clear that complement is not a single entity; instead, it is a system that is activated by multiple pathways involving a complex cascade of protease activation. It also became apparent that the system is tightly regulated at various activation steps and lack of regulation results in disease. The characterization of effectors revealed that the system targets pathogens by marking them as ‘non-self’ by opsonins and such tagging of pathogens results in their phagocytosis through complement receptors and lysis owing to loss of their membrane integrity. The complement-activated serum was also shown to have ‘anaphylatoxin’ activity, which was later found to be associated with smooth muscle contraction, enhanced vascular permeability and recruitment of white blood cells [5]. As expected, this activity was demonstrated to contribute to containing localized infections [6]. In the later part of the century, it also became clear that the complement system participates in enhancing pathogen-specific B and T cell responses [7,8][7][8] as well as induction of an antiviral state [9].
Thus, it is evident from the above account that complement is a formidable defense against infectious agents, including viruses, as it can not only act directly on pathogens but also enhance innate and adaptive immunity against them. The evolution of viruses, nevertheless, is shaped by constant host-induced pressures [10]. Hence it is not surprising that viruses have developed mechanisms to thwart complement attack. These include encoding molecules to subvert the complement system, acquisition of host complement regulatory proteins, employment of complement receptors for cellular entry and upregulation of host complement regulatory proteins on the infected cells.
2. Historical Perspective
The first virally encoded molecule that was identified as a complement regulator was herpes simplex virus type 1 (HSV-1) glycoprotein C (gC-1) (Figure 1). In 1982, Harvey Friedman and his group from the University of Pennsylvania observed that human endothelial cells infected with HSV-1 express a C3 receptor [11]. Later, they identified this molecule as gC-1, which has no homology to the known complement receptors [12]. Detailed studies revealed that the molecule is not a receptor; instead, it functions as a C3 regulator [13]. Such mechanism of complement regulation, however, was not unique to HSV-1 as a structurally similar glycoprotein present on HSV-2 could also inhibit complement [14].
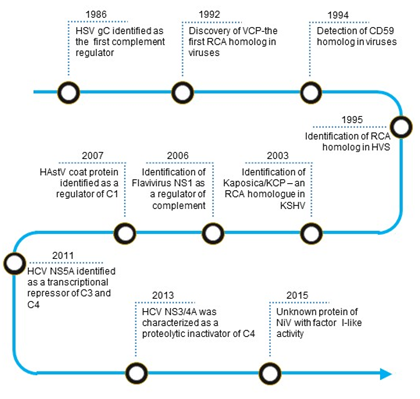
Figure 1. Timeline of identification of virally encoded complement regulators. Abbreviations: HSV gC, Herpes simplex virus glycoprotein C; VCP, Vaccinia virus complement control protein; RCA, regulator of complement activation; HVS, Herpesvirus saimiri; KSHV, Kaposi’s sarcoma-associated herpesvirus; Kaposica, KSHV inhibitor of complement activation; KCP, KSHV complement control protein; NS1, Non-structural protein; HAstV coat protein, Human astrovirus coat protein; HCV NS5A, Hepatitis C virus non-structural 5A protein; HCV NS3/4A, Hepatitis C virus non-structural 3/4A protease; NiV, Nipah virus.
In humans, the major complement regulators belong to a gene family termed the regulators of complement activation (RCA). These regulators are exclusively formed by concatenated bead-like domains—the complement control protein (CCP) domains [15]. A homolog of the RCA gene family proteins was first reported in the vaccinia virus in 1988 by Kotwal and Moss [16]. Its biochemical characterization established that the protein essentially functions as human regulators [17]. In a quest to identify such RCA homologs in other viruses, various laboratories examined viral genomes for the presence of sequence homologs. They discovered that apart from poxviruses, RCA homologs also exist in gammaherpesviruses, including Kaposi’s sarcoma-associated herpesvirus [18–20] [18][19][20]. We now know that functionality in these proteins is dictated by the presence of spatially conserved motifs [21].
The membrane attack complex (MAC) is a critical effector of the complement system that can form pores on the plasma membrane of the target cells. Consequently, its formation on the viral envelope results in loss of viral integrity. In 1992, based on sequence similarity, a group at the Institut fur Klinische und Molekulare Virologie, Erlangen discovered that Herpesvirus saimiri (HVS) encodes a homolog of human CD59 [22]. Later, the laboratories of Stephen Squinto as well as Peter Lachmann, showed that the protein indeed has an ability to block complement-mediated cytolysis [23,24][23][24].
Besides RCA and CD59 homologs, various other non-structural viral proteins are known to mediate complement evasion. Among these, non-structural protein 1 (NS1) of flaviviruses is known to subvert the complement system. In the 2000s, the laboratories of John Atkinson and Michael Diamond at the Washington University in St. Louis demonstrated that secreted hexamer of NS1 in particular recruits complement regulators such as factor H and C4b-binding protein onto the infected cells and protect them from complement attack [25,26][25][26]. Additionally, the hexamer was also shown to antagonize complement C4 in solution [27]. Ranjit Ray’s laboratory at St. Louis University School of Medicine showed that the non-structural proteins of hepatitis C virus subvert complement by utilizing different mechanisms. NS5A transcriptionally downregulates the expression of complement components like C3, C4, and C9 which participate in complement activation, while NS3/4A proteolytically inactivates C4 [28–30][28][29][30].
The complement proteins C1q and mannose-binding lectin (MBL) are pattern recognition molecules which deftly recognize viruses. In the 2010s, it became clear that viruses have also developed mechanisms to block the interaction of these pattern recognition molecules with viruses. The laboratory of Neel Krishna at the Virginia Medical School showed that the astrovirus capsid protein interacts with C1q and inhibits the classical pathway of complement activation likely due to the displacement of protease tetramer C1s-C1r-C1r-C1s [31]. The capsid protein was also shown to interact with MBL and inhibit the lectin pathway [31]. In addition to the capsid protein of astroviruses, the M1 protein of Influenza A virus was also shown to interact with C1q and inhibit the classical complement-mediated neutralization of the virus [32].
Tagging of viruses with C3b is critical for complement-mediated inactivation and clearance. Hence, efficient inactivation of virus tagged C3b is central for viral protection. Recent reports show that RNA viruses like Nipah and Chikungunya display factor I-like activity which can mediate C3b inactivation [33] [33]. This mechanism appears to be unique to viruses as no other pathogen has been shown to exhibit factor I-like activity.
3. Viral Complement Regulators
Virus replication is initiated after its attachment to a cell, which is then followed by its cellular entry, uncoating, replication, assembly, and release. Thus, before its entry and after the release, the virus is in the extracellular milieu, while during the other stages, it is inside a cell. An effective innate immune defense, therefore, must target both phases of the virus life cycle. Notably, all the major complement pathways—classical, alternative, and lectin—are potent in targeting cell-free viral particles as well as the virus-infected cells [34–37][34][35][36][37]. Moreover, as stated above, the system also boosts other antiviral innate [9] and acquired immune responses [7,8] to limit the viral infection. Studies on the complement subversion mechanisms of viruses have shown that viruses employ structural (i.e., components of the viral particles) as well as non-structural proteins to subvert these responses (Figure 2 and Table 1).
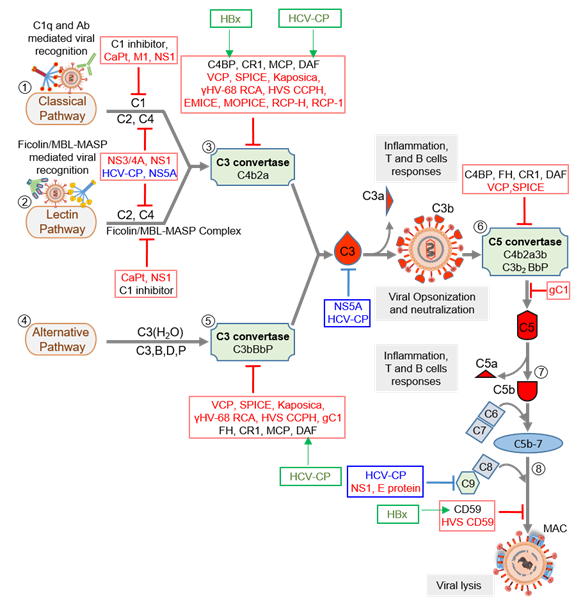
Figure 2. Complement activation and its regulation by host regulators and virally encoded molecules. Viruses can activate the host complement system by three major pathways: classical pathway (CP), lectin pathway (LP), and alternative pathway (AP). (1) In CP activation, viruses are known to be recognized by C1q and antibody. (2) In LP activation, viruses are recognized by the Ficolin/MBL-MASP complex. (3) Recognition is followed by activation of C1 and Ficolin/MBL complexes, in CP and LP respectively, which results in the cleavage of C4 and C2 and formation of C3 convertase C4b2a. The convertase then cleaves C3 into C3a and C3b. The latter opsonizes viral particles. (4) In AP, viruses are recognized directly by C3b molecules, which are generated by the initial C3 convertase C3(H2O)Bb. (5) The surface-bound C3b molecules then trigger the formation of C3 convertase C3bBb with the help of factors B and D, which is stabilized by properdin (P). The C3 convertases formed then cleaves more C3 to opsonize viral particles. (6) When C3b is attached to the preformed C3 convertase (C4b2a or C3bBbP), it is converted into C5 convertase (C4b2a3b or C3b2BbP) which is capable of cleaving C5 into C5a and C5b. (7) The newly formed C5b combines C6 and C7 to form a C5b-7 trimer that can attach to the viral surface. (8) Binding of the trimer to C8 and C9 followed by polymerization of C9 results in the formation of the membrane attack complex (MAC) that induces virolysis. These pathways are regulated at various steps by host complement regulators like C1 inhibitor, C4b-binding protein (C4BP), complement receptor 1 (CR1; CD35), membrane cofactor protein (MCP; CD46), decay-accelerating factor (DAF; CD55), factor H (FH) and CD59. Viral complement regulators that target complement proteins, enzymes, and complexes are shown in red text, whereas those that inhibit complement proteins’ expression are shown in blue text. Some viral complement regulators enhance the expression of host complement regulators. These are identified in green text, and green arrows mark the regulator they upregulate. Abbreviations: CaPt, Astrovirus capsid protein; M1, Influenza virus matrix protein 1; NS1, Flavivirus non-structural protein 1; NS3/4A, Hepatitis C virus non-structural 3/4A protease; HCV-CP, Hepatitis C virus core protein; NS5A, Hepatitis C virus non-structural 5A protein; VCP, Vaccinia virus complement control protein; SPICE, Smallpox inhibitor of complement enzymes; Kaposica, KSHV inhibitor of complement activation; γHV-68 RCA, Murine gammaherpesvirus 68 regulator of complement activation; HVS CCPH, Herpesvirus saimiri complement control protein homolog; EMICE, Ectromelia virus inhibitor of complement enzymes; MOPICE, Monkeypox inhibitor of complement enzymes; RCP-H, Rhesus rhadinovirus complement control protein H; RCP-1, Rhesus rhadinovirus complement control protein-1; gC1, Herpes simplex virus glycoprotein C-1; E protein, Zika virus E protein, HVS CD59, Herpesvirus saimiri CD59; HBx, Hepatitis B virus X protein.
Table 1. Viruses and their complement regulators.
|
Virus Family |
Virus |
Complement Evasion Protein |
Target |
Reference |
|
Herpesviridae |
Herpes simplex virus |
Glycoprotein C-1 |
AP C3 convertase and C3b |
[13,38] |
|
Herpesvirus saimiri |
CD59 homolog |
C5b-8 and C5b-9 |
[23,24] |
|
|
HVS CCPH |
CP/LP and AP C3 convertase |
[39,40] |
||
|
Kaposi sarcoma-associated herpesvirus |
Kaposica |
CP/LP and AP C3 convertase |
[19,20] |
|
|
Rhesus rhadinovirus |
RCP-H and RCP-1 |
CP/LP and AP C3 convertase |
[41,42] |
|
|
Murine gammaherpesvirus 68 |
γHV68 RCA protein |
CP/LP and AP C3 convertase |
[43] |
|
|
Astroviridae |
Astrovirus |
CaPt |
C1q and MBL |
[31,44] |
|
Orthomyxoviridae |
Influenza virus |
M1 |
C1q |
[32] |
|
Paramyxoviridae |
Nipah virus |
Unknown protein with factor I like activity |
C3b |
[33] |
|
Togaviridae |
Chikungunya virus |
Unknown protein with factor I like activity |
C3b |
[45] |
|
Flaviviridae |
Hepatitis C virus |
Core protein |
C3, C4 and C9 genes |
[28–30] |
|
NS3/4A |
C4 |
[46] |
||
|
NS5A |
C3 and C4 genes |
[28,47] |
||
|
West Nile virus and Dengue virus |
NS1 |
C4 and C9 |
[26,27] |
|
|
Zika virus |
E protein |
C5b-6, C8 and C9 |
[48] |
|
|
Hepadnaviridae |
Hepatitis B virus |
HBx protein |
CD59 and C4BP genes |
[49,50] |
|
Poxviridae |
Variola virus |
SPICE |
CP/LP and AP C3/C5 convertase |
[51,52] |
|
Vaccinia virus |
VCP |
CP/LP and AP C3/C5 convertase |
[17,53,54] |
|
|
Cowpox virus |
IMP |
Unknown |
[55] |
|
|
Monkeypox virus |
MOPICE |
CP/LP C3 convertase |
[56] |
|
|
Ectromelia virus |
EMICE |
CP/LP C3 convertase |
[57] |
CP—Classical pathway, AP—Alternative pathway, LP—Lectin pathway.
References
- Wasik, B.R.; Turner, P.E. On the Biological Success of Viruses. Annu. Rev Microbiol. 2013, 67, 519–541.
- Doherty, M.; Robertson, M.J. Some Early Trends in Immunology. Trends Immunol. 2004, 25, 623–631.
- Lachmann, P. Complement Before Molecular Biology. Mol. Immunol. 2006, 43, 496–508.
- Bordet, J.; Gengou, O. Sur L’Existences de Substances Sensibilisatrices Dans la Plupart des Serums Anti-Microbiens. Annales de I'Institut Pasteur 1901, 15, 289–302.
- Ember, J.A.; Jagels, M.A.; Hugli, T.E. Characterization of Complement Anaphylatoxins and Their Biological Responses. In The Human Complement System in Health and Disease; Volanakis, J.E., Frank, M.M., Eds.; Marcel Dekker, Inc.: New York, NY, USA, 1998; Chapter 11.
- Hopken, U.E.; Lu, B.; Gerard, N.P.; Gerard, C. The C5a Chemoattractant Receptor Mediates Mucosal Defence to Infection. Nature 1996, 383, 86–89.
- Fearon, D.T.; Carroll, M.C. Regulation of B Lymphocyte Responses to Foreign and Self-Antigens by the CD19/CD21 Com-plex. Annu. Rev Immunol. 2000, 18, 393–422.
- West, E.E.; Kolev, M.; Kemper, C. Complement and the Regulation of T Cell Responses. Annu. Rev Immunol. 2018, 36, 309–338.
- Tam, J.C.; Bidgood, S.R.; McEwan, W.A.; James, L.C. Intracellular Sensing of Complement C3 Activates Cell Autonomous Immunity. Science 2014, 345, 1256070.
- Paterson, S.; Vogwill, T.; Buckling, A.; Benmayor, R.; Spiers, A.J.; Thomson, N.R.; Quail, M.; Smith, F.; Walker, D.; Libberton, B.; et al. Antagonistic Coevolution Accelerates Molecular Evolution. Nature 2010, 464, 275–278.
- Cines, D.B.; Lyss, A.P.; Bina, M. Fc and C3 Receptors Induced by Herpes Simplex Virus on Cultured Human Endothelial Cells. J. Clin. Invest. 1982, 69, 123–128.
- Friedman, H.M.; Glorioso, J.C.; Cohen, G.H.; Hasting, J.C.; Harris, S.L.; Eisenberg, R.J. Binding of Complement Component C3b to Glycoprotein GC of Herpes Simplex Virus Type 1: Mapping of GC-Binding Sites and Demonstration of Conserved C3b Binding in Low-Passage Clinical Isolates. J. Virol. 1986, 60, 470–475.
- Fries, L.F.; Friedman, H.M.; Cohen, G.H.; Eisenberg, R.J.; Hammer, C.H.; Frank, M.M. Glycoprotein C of Herpes Simplex Virus 1 Is an Inhibitor of the Complement Cascade. J. Immunol. 1986, 137, 1636–1641.
- McNearney, T.A.; Odell, C.; Holers, V.M.; Spear, P.G.; Atkinson, J.P. Herpes Simplex Virus Glycoproteins GC-1 and GC-2 Bind to the Third Component of Complement and Provide Protection Against Complement-Mediated Neutralization of Viral Infectivity. J. Exp. Med. 1987, 166, 1525–1535.
- Panwar, H.S.; Ojha, H.; Ghosh, P.; Barage, S.H.; Raut, S.; Sahu, A. Molecular Engineering of an Efficient Four-Domain DAF-MCP Chimera Reveals the Presence of Functional Modularity in RCA Proteins. Proc. Natl. Acad. Sci. USA 2019, 116, 9953–9958.
- Kotwal, G.J.; Moss, B. Vaccinia Virus Encodes a Secretory Polypeptide Structurally Related to Complement Control Proteins. Nature 1988, 335, 176–178.
- Kotwal, G.J.; Isaacs, S.N.; Mckenzie, R.; Frank, M.M.; Moss, B. Inhibition of the Complement Cascade by the Major Secretory Protein of Vaccinia Virus. Science 1990, 250, 827–830.
- Moore, P.S.; Chang, Y. Kaposi’s Sarcoma-Associated Herpesvirus Immunoevasion and Tumorigenesis: Two Sides of the Same Coin? Annu. Rev. Microbiol. 2003, 57, 609–639.
- Mullick, J.; Bernet, J.; Singh, A.K.; Lambris, J.D.; Sahu, A. Kaposi’s Sarcoma-Associated Herpesvirus (Human Herpesvirus-8) Open Reading Frame 4 Protein (Kaposica) Is a Functional Homolog of Complement Control Proteins. J. Virol. 2003, 77, 3878–3881.
- Spiller, O.B.; Blackbourn, D.J.; Mark, L.; Proctor, D.G.; Blom, A.M. Functional Activity of the Complement Regulator Encoded by Kaposi’s Sarcoma-Associated Herpesvirus. J. Biol. Chem. 2003, 278, 9283–9289.
- Ojha, H.; Ghosh, P.; Singh, P.H.; Shende, R.; Gondane, A.; Mande, S.C.; Sahu, A. Spatially Conserved Motifs in Complement Control Protein Domains Determine Functionality in Regulators of Complement Activation-Family Proteins. Commun. Biol. 2019, 2, 290.
- Albrecht, J.C.; Fleckenstein, B. New Member of the Multigene Family of Complement Control Proteins in Herpesvirus Saimiri. J. Virol. 1992, 66, 3937–3940.
- Rother, R.P.; Rollins, S.A.; Fodor, W.L.; Albrecht, J.C.; Setter, E.; Fleckenstein, B.; Squinto, S.P. Inhibition of Comple-ment-Mediated Cytolysis by the Terminal Complement Inhibitor of Herpesvirus Saimiri. J. Virol. 1994, 68, 730–737.
- Bramley, J.C.; Davies, A.; Lachmann, P.J. Herpesvirus Saimiri CD59—Baculovirus Expression and Characterisation of Com-plement Inhibitory Activity. Biochem. Soc. Trans. 1997, 25, 354S.
- Avirutnan, P.; Hauhart, R.E.; Somnuke, P.; Blom, A.M.; Diamond, M.S.; Atkinson, J.P. Binding of Flavivirus Nonstructural Protein NS1 to C4b Binding Protein Modulates Complement Activation. J. Immunol. 2011, 187, 424–433.
- Conde, J.N.; da Silva, E.M.; Allonso, D.; Coelho, D.R.; Andrade, I.D.; de Medeiros, L.N.; Menezes, J.L.; Barbosa, A.S.; Mo-hana-Borges, R. Inhibition of the Membrane Attack Complex by Dengue Virus NS1 Through Interaction with Vitronectin and Terminal Complement Proteins. J. Virol. 2016, 90, 9570–9581.
- Avirutnan, P.; Fuchs, A.; Hauhart, R.E.; Somnuke, P.; Youn, S.; Diamond, M.S.; Atkinson, J.P. Antagonism of the Comple-ment Component C4 by Flavivirus Nonstructural Protein NS1. J. Exp. Med. 2010, 207, 793–806.
- Banerjee, A.; Mazumdar, B.; Meyer, K.; Di Bisceglie, A.M.; Ray, R.B.; Ray, R. Transcriptional Repression of C4 Complement by Hepatitis C Virus Proteins. J. Virol. 2011, 85, 4157–4166.
- Mazumdar, B.; Kim, H.; Meyer, K.; Bose, S.K.; Di Bisceglie, A.M.; Ray, R.B.; Diamond, M.S.; Atkinson, J.P.; Ray, R. Hepatitis C Virus Infection Upregulates CD55 Expression on the Hepatocyte Surface and Promotes Association with Virus Particles. J. Virol. 2013, 87, 7902–7910.
- Kim, H.; Meyer, K.; Di Bisceglie, A.M.; Ray, R. Hepatitis C Virus Suppresses C9 Complement Synthesis and Impairs Mem-brane Attack Complex Function. J. Virol. 2013, 87, 5858–5867.
- Hair, P.S.; Gronemus, J.Q.; Crawford, K.B.; Salvi, V.P.; Cunnion, K.M.; Thielens, N.M.; Arlaud, G.J.; Rawal, N.; Krishna, N.K. Human Astrovirus Coat Protein Binds C1q and MBL and Inhibits the Classical and Lectin Pathways of Complement Activation. Mol. Immunol. 2010, 47, 792–798.
- Zhang, J.; Li, G.; Liu, X.; Wang, Z.; Liu, W.; Ye, X. Influenza A Virus M1 Blocks the Classical Complement Pathway Through Interacting with C1qA. J. Gen. Virol. 2009, 90, 2751–2758.
- Johnson, J.B.; Borisevich, V.; Rockx, B.; Parks, G.D. A Novel Factor I Activity in Nipah Virus Inhibits Human Complement Pathways Through Cleavage of C3b. J. Virol. 2015, 89, 989–998.
- Agrawal, P.; Sharma, S.; Pal, P.; Ojha, H.; Mullick, J.; Sahu, A. The Imitation Game: A Viral Strategy to Subvert the Com-plement System. FEBS Lett. 2020, 594, 2518–2542.
- Perrin, L.H.; Joseph, B.S.; Cooper, N.R.; Oldstone, M.B. Mechanism of Injury of Virus-Infected Cells by Antiviral Antibody and Complement: Participation of IgG, F(Ab’)2, and the Alternative Complement Pathway. J. Exp. Med. 1976, 143, 1027–1041.
- Sissons, J.G.; Oldstone, M.B.; Schreiber, R.D. Antibody-Independent Activation of the Alternative Complement Pathway by Measles Virus-Infected Cells. Proc. Natl. Acad. Sci. USA 1980, 77, 559–562.
- Liu, J.; Ali, M.A.; Shi, Y.; Zhao, Y.; Luo, F.; Yu, J.; Xiang, T.; Tang, J.; Li, D.; Hu, Q.; et al. Specifically Binding of L-Ficolin to N-Glycans of HCV Envelope Glycoproteins E1 and E2 Leads to Complement Activation. Cell Mol. Immunol. 2009, 6, 235–244.