The MUTYH gene is located on the short arm of chromosome 1 (1p34.1) [15] and encodes instructions for the MYH glycosylase enzyme.
- MUTYH
- ovarian cancer
1. MUTYH Gene Function
The MUTYH gene is located on the short arm of chromosome 1 (1p34.1) [15] [1] and encodes instructions for the MYH glycosylase enzyme. This enzyme repairs DNA damage via a base excision repair mechanism. Many different carcinogens damage DNA, including reactive oxygen species, alkylating agents, DNA cross-linking agents, and radiation. Although MUTYH performs base excision repair and initiation of apoptosis in response to DNA damage from alkylation [2] [16] and ultraviolet radiation [17][3], its primary function is to repair oxidative DNA damage [18][4].
Following stimulation from the MSH6 component of the MSH2/MSH6 heterodimer, MUTYH identifies and binds a mismatch of 8-hydroxyguanine (8-oxoG):A [19][5]. Myh glycosylase then excises the mismatched adenine, preventing inappropriate G:C > T:A transversions in subsequent rounds of DNA replication [15,18,20,21][1][4][6][7]. Removal of the inappropriate base-pairing generates an apurinic/apyrimidinic (AP) site [22][8]. A physical connection between MYH and AP endonuclease I (APE1) enables prompt action by APE1 to nick the DNA phosphodiester backbone, causing a single-strand DNA break [9] [23] (Figure 1).
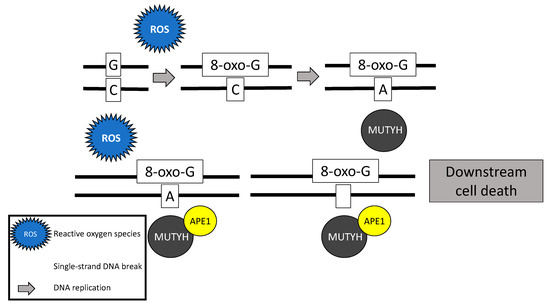
Figure 1. Base excision repair mechanism of the MutY homolog (MUTYH) in response to oxidative damage.
In addition to its well-known role in base excision repair, MUTYH also plays a role in rapid DNA damage response by inducing cell death [21,24,25][7][10][11]. Oka and colleagues (2008) demonstrated that single-strand mitochondrial DNA breaks performed by MYH’s base excision repair mechanism result in cell death via calpain activation [25][11], which is a p53 independent process [24,25][10][11]. Alternatively, nuclear DNA breaks induced by MYH glycosylase result in cell death via a poly-adenosine diphosphate (ADP) ribose polymerase (PARP) signaling pathway [25][11], which is mediated by the tumor suppressor protein, p53 [10] [24] and the mismatch repair gene MLH1 [26][12]. PARP senses and binds to single-strand DNA breaks, ribosylates itself and other cellular proteins, and signals apoptosis-inducing factor to translocate from the mitochondria to the nucleus, resulting in cell death [27][13].
Another method by which MUTYH responds to rapid DNA damage involves activation and phosphorylation of the tumor suppressor gene, checkpoint kinase 1 (CHEK1) via ataxia telangiectasia and rad3-related protein (ATR), another tumor suppressor gene [28,29] [14][15]. When activated, CHEK1 activates cell cycle checkpoint processes prompting DNA repair and/or apoptosis [30][16]. Following exposure to DNA damaging agents, Hahm and colleagues (2011) found less CHEK1 and ATR phosphorylation in knockdown MUTYH cells than in wild-type cells [28][14]. When this pathway is altered, the activity of checkpoint kinase 2 (CHEK2) increases [28][14].
2. MUTYH Germline Mutations
The estimated prevalence of a heterozygous MUTYH germline mutation is 1–2% [17][18] [31,32] and homozygous germline mutations is 0.012% [33,34][19][20]. Most pathogenic MUTYH mutations are missense [18,35][4][21], including Y179C (c.536A > G; previously Y165C (c.494A4G)) and G396D (c.1187G > A; previously G382D (c.1145G4A)). These mutations account for approximately 70% of all pathogenic mutations in Western populations [31,35][17][21] likely due to a genetic founder effect [21][7]. Although approximately 18% of all ovarian cancers are the result of an inherited predisposition [2], it is unknown how many of these ovarian cancers are directly attributable to biallelic MUTYH mutations. Consistent with the estimated prevalence of carrying a heterozygous MUTYH germline mutation [31[17][18],32], monoallelic germline MUTYH mutations have been identified in 1.9% of ovarian cancer patients [36][22].
3. MUTYH Somatic Mutations
Somatic MUTYH mutations are increasingly described in the literature and are noted to be present in 3.3% of all tumors curated by the Catalogue of Somatic Mutations in Cancer (COSMIC) database [37][23]. Similar to germline mutations, most (56.7%) mutations are missense. Over 400 different somatic MUTYH mutations have been curated by COSMIC and the most frequently identified is c157 + 30A > G, occurring 6% of the time [37] [23]. Somatic MUTYH mutations have been described in sporadic colorectal cancer and may occur concurrent to somatic adenomatous polyposis coli (APC) gene mutations [38][24]. Furthermore, MUTYH somatic mutations have been described in Lynch syndrome patients and in patients whose tumors demonstrate mismatch repair deficiency but carry no mismatch repair germline mutation [39] [25]. Outside of colon cancer, somatic MUTYH mutations have also been described in breast cancers [40,41][26][27] +and in 0.24% of ovarian cancer samples [37][23].
4. MUTYH Mutation and Mechanism for Oncogenesis
While somatic and biallelic MUTYH mutations are associated with an increased risk of ovarian cancer, the majority of mechanistic studies have been performed in colon cancer model systems. Due to the critical role MUTYH plays in base excision repair, DNA with functional loss of MUTYH leads to an excess of 8-oxoguanine (8-oxo-G) which results in inappropriate G:C > T:A transversions in MAP colorectal tumors [4][6][28] [18,20,42] (Figure 2). Without active MUTYH activity, 8-oxoguanine glycosylase (OGG1), a DNA glycosylase enzyme involved in base excision repair, may identify and remove 8-oxo-G in an inappropriate 8-oxo-G:A pairing resulting in another mechanism for incorrect G:C > T:A transversions; competition between MUTYH and OGG1 has demonstrated in a mouse model [21,43][7][29].
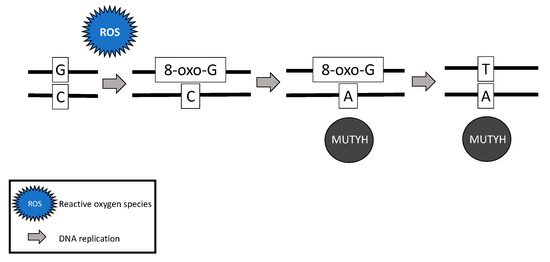
Figure 2. Failure of MUTYH’s base excision repair mechanism and resultant G > T transversion.
The somatic G > T transversions seen in MAP tumors are likely the result of unrepaired DNA damage from reactive oxygen species [18][4]. Over time, increasing numbers of G > T transversions in somatic tissues are likely to impact various oncogenes and tumor suppressor genes and lead to cancer [21,44,45][7][30][31]. For example, MAP colon tumors demonstrate a high prevalence of somatic APC [6][28] [20,42] and Kirsten rat sarcoma (KRAS) [42][28] G > T transversions. MAP colon tumors have also demonstrated somatic G > T transversions in mismatch repair genes MLH1 [46][32], MSH2, and MSH6 [47][33], resulting in microsatellite instability and mimicking Lynch syndrome [46,47][32][33].
In addition to carcinogenesis resulting from failed base excision repair, tumorigenesis may result from the failure of damaged cells to undergo apoptosis [24][10]. Oka and colleagues (2014) demonstrated that MUTYH knockdown colorectal cancer cells exposed to oxidative stress failed to undergo PARP-mediated apoptosis, resulting in aberrant cell growth and risk for tumorigenesis [24][10]. The tumor suppressor protein, p53 [26][12], regulates this process: the same aberrant cell growth was identified with either p53 deficiency or MUTYH knockout [24][10]. Although no studies have investigated the effect of mutant MUTYH in ovarian cancer cells, the mechanism for ovarian cancer oncogenesis is likely similar to that of colorectal cancer carcinogenesis as both of these cancers are established in the MAP phenotype. Furthermore, impaired MUTYH function in human keratinocyte HaCaT and human embryonic kidney HEK293 cells fail to phosphorylate ATR/CHEK1 [28][14]. Failure to prompt DNA repair and/or apoptosis [30] [16] may shift cell checkpoint messages to CHEK2 control. Although there is significant overlap in CHEK1 and CHEK2 functions [28][14], without CHEK1 control, cells exposed to DNA damaging ionizing radiation fail to arrest in the G2 phase, leaving the possibility of survival and replication of damaged DNA and another potential mechanism for tumorigenesis [48][34]. Although these findings suggest carcinogenic plausibility, future research may test this hypothesis in ovarian cancer cell lines.
References
- Vogt, S.; Jones, N.; Christian, D.; Engel, C.; Nielsen, M.; Kaufmann, A.; Steinke, V.; Vasen, H.F.; Propping, P.; Sampson, J.R.; et al. Expanded extracolonic tumor spectrum in MUTYH-associated polyposis. Gastroenterology 2009, 137, 1976–1985.e10, doi:10.1053/j.gastro.2009.08.052.
- United States National Library of Medicine. MUTYH Gene. Available online: https://medlineplus.gov/genetics/gene/mutyh/ (accessed on 28 July 2020).
- Fry, R.C.; Svensson, J.P.; Valiathan, C.; Wang, E.; Hogan, B.J.; Bhattacharya, S.; Bugni, J.M.; Whittaker, C.A.; Samson, L.D. Genomic predictors of interindividual differences in response to DNA damaging agents. Genes Dev. 2008, 22, 2621–2626, doi:10.1101/gad.1688508.
- Mazouzi, A.; Battistini, F.; Moser, S.C.; Ferreira da Silva, J.; Wiedner, M.; Owusu, M.; Lardeau, C.H.; Ringler, A.; Weil, B.; Neesen, J.; et al. Repair of UV-induced DNA damage independent of nucleotide excision repair is masked by MUTYH. Mol. Cell 2017, 68, 797–807.e7, doi:10.1016/j.molcel.2017.10.021.
- Molatore, S.; Russo, M.T.; D’Agostino, V.G.; Barone, F.; Matsumoto, Y.; Albertini, A.M.; Minoprio, A.; Degan, P.; Mazzei, F.; Bignami, M.; et al. MUTYH mutations associated with familial adenomatous polyposis: Functional characterization by a mammalian cell-based assay. Hum. Mutat. 2010, 31, 159–166, doi:10.1002/humu.21158.
- Gu, Y.; Parker, A.; Wilson, T.M.; Bai, H.; Chang, D.Y.; Lu, A.L. Human MutY homolog, a DNA glycosylase involved in base excision repair, physically and functionally interacts with mismatch repair proteins human MutS homolog 2/human MutS homolog 6. J. Biol. Chem. 2002, 277, 11135–11142, doi:10.1074/jbc.M108618200.
- Al-Tassan, N.; Chmiel, N.H.; Maynard, J.; Fleming, N.; Livingston, A.L.; Williams, G.T.; Hodges, A.K.; Davies, D.R.; David, S.S.; Sampson, J.R.; et al. Inherited variants of MYH associated with somatic G:C-->T:A mutations in colorectal tumors. Nat. Genet. 2002, 30, 227–232, doi:10.1038/ng828.
- Raetz, A.G.; David, S.S. When you’re strange: Unusual features of the MUTYH glycosylase and implications in cancer. DNA Repair 2019, 80, 16–25, doi:10.1016/j.dnarep.2019.05.005.
- Jansson, K.; Alao, J.P.; Viktorsson, K.; Warringer, J.; Lewensohn, R.; Sunnerhagen, P. A role for Myh1 in DNA repair after treatment with strand-breaking and crosslinking chemotherapeutic agents. Environ. Mol. Mutagen. 2013, 54, 327–337, doi:10.1002/em.21784.
- Luncsford, P.J.; Manvilla, B.A.; Patterson, D.N.; Malik, S.S.; Jin, J.; Hwang, B.J.; Gunther, R.; Kalvakolanu, S.; Lipinski, L.J.; Yuan, W.; et al. Coordination of MYH DNA glycosylase and APE1 endonuclease activities via physical interactions. DNA Repair (Amst) 2013, 12, 1043–1052, doi:10.1016/j.dnarep.2013.09.007.
- Oka, S.; Leon, J.; Tsuchimoto, D.; Sakumi, K.; Nakabeppu, Y. MUTYH, an adenine DNA glycosylase, mediates p53 tumor suppression via PARP-dependent cell death. Oncogenesis 2014, 3, e121, doi:10.1038/oncsis.2014.35.
- Oka, S.; Leon, J.; Tsuchimoto, D.; Sakumi, K.; Nakabeppu, Y. Erratum: MUTYH, an adenine DNA glycosylase, mediates p53 tumor suppression via PARP-dependent cell death. Oncogenesis 2015, 4, e142, doi:10.1038/oncsis.2015.4.
- Oka, S.; Ohno, M.; Tsuchimoto, D.; Sakumi, K.; Furuichi, M.; Nakabeppu, Y. Two distinct pathways of cell death triggered by oxidative damage to nuclear and mitochondrial DNAs. EMBO J. 2008, 27, 421–432, doi:10.1038/sj.emboj.7601975.
- McDaid, J.R.; Loughery, J.; Dunne, P.; Boyer, J.C.; Downes, C.S.; Farber, R.A.; Walsh, C.P. MLH1 mediates PARP-dependent cell death in response to the methylating agent N-methyl-N-nitrosourea. Br. J. Cancer 2009, 101, 441–451, doi:10.1038/sj.bjc.6605186.
- Fatokun, A.A.; Dawson, V.L.; Dawson, T.M. Parthanatos: Mitochondrial-linked mechanisms and therapeutic opportunities. Br. J. Pharmacol. 2014, 171, 2000–2016, doi:10.1111/bph.12416.
- Hahm, S.H.; Park, J.H.; Ko, S.I.; Lee, Y.R.; Chung, I.S.; Chung, J.H.; Kang, L.W.; Han, Y.S. Knock-down of human MutY homolog (hMYH) decreases phosphorylation of checkpoint kinase 1 (Chk1) induced by hydroxyurea and UV treatment. BMB Rep. 2011, 44, 352–357, doi:10.5483/BMBRep.2011.44.5.352.
- Zhao, H.; Piwnica-Worms, H. ATR-mediated checkpoint pathways regulate phosphorylation and activation of human Chk1. Mol. Cell Biol. 2001, 21, 4129–4139, doi:10.1128/MCB.21.13.4129-4139.2001.
- Patil, M.; Pabla, N.; Dong, Z. Checkpoint kinase 1 in DNA damage response and cell cycle regulation. Cell Mol. Life Sci. 2013, 70, 4009–4021, doi:10.1007/s00018-013-1307-3.
- Nielsen, M.; Infante, E.; Brand, R. MUTYH polyposis. In GeneReviews [Internet]; Adam, M.P., Ardinger, H.H., Pagon, R.A. Wallace, S.E., Eds.; [Updated 2019]; University of Washington: Seattle, WA, USA, 2012. Available online: https://www.ncbi.nlm.nih.gov/books/NBK107219/# (accessed on 30 July 2020).
- Samadder, N.J.; Riegert-Johnson, D.; Boardman, L.; Rhodes, D.; Wick, M.; Okuno, S.; Kunze, K.L.; Golafshar, M.; Uson, P.L.S.; Mountjoy, L.; et al. Comparison of universal genetic testing vs guideline-directed targeted testing for patients with he-reditary cancer syndrome. JAMA Oncol. 2020, doi:10.1001/jamaoncol.2020.6252.
- Win, A.K.; Hopper, J.L.; Jenkins, M.A. Association between monoallelic MUTYH mutation and colorectal cancer risk: A me-ta-regression analysis. Fam. Cancer 2011, 10, 1–9, doi:10.1007/s10689-010-9399-5.
- Win, A.K.; Jenkins, M.A.; Dowty, J.G.; Antoniou, A.C.; Lee, A.; Giles, G.G.; Buchanan, D.D.; Clendenning, M.; Rosty, C.; Ahnen, D.J.; et al. Prevalence and penetrance of major genes and polygenes for colorectal cancer. Cancer Epidemiol. Bi-omarkers Prev. 2017, 26, 404–412, doi:10.1158/1055-9965.EPI-16-0693.
- Cheadle, J.P.; Sampson, J.R. MUTYH-associated polyposis--from defect in base excision repair to clinical genetic testing. DNA Repair (Amst) 2007, 6, 274–279, doi:10.1016/j.dnarep.2006.11.001.
- Minion, L.E.; Dolinsky, J.S.; Chase, D.M.; Dunlop, C.L.; Chao, E.C.; Monk, B.J. Hereditary predisposition to ovarian cancer, looking beyond BRCA1/BRCA2. Gynecol. Oncol. 2015, 137, 86–92, doi:10.1016/j.ygyno.2015.01.537.
- Catalogue of Somatic Mutations in Cancer (COSMIC): MUTYH Gene. Available online: https://cancer.sanger.ac.uk/cosmic/gene/analysis?ln=MUTYH (accessed on 25 August 2020).
- Bougatef, K.; Marrakchi, R.; Kourda, N.; Ben Lahely, Y.B.; Jileni, S.B.; El Khil, H.K.; Soubrier, F.; Ben Ammar Elgaaied, A. Somatic mutation of MutYH in Tunisian patients with sporadic colorectal cancer. J. Clin. Lab. Anal. 2007, 21, 372–374, doi:10.1002/jcla.20198.
- Vargas-Parra, G.M.; Gonzalez-Acosta, M.; Thompson, B.A.; Gomez, C.; Fernandez, A.; Damaso, E.; Pons, T.; Morak, M.; Del Valle, J.; Iglesias, S.; et al. Elucidating the molecular basis of MSH2-deficient tumors by combined germline and somatic anal-ysis. Int. J. Cancer 2017, 141, 1365–1380, doi:10.1002/ijc.30820.
- Thibodeau, M.L.; Zhao, E.Y.; Reisle, C.; Ch'ng, C.; Wong, H.L.; Shen, Y.; Jones, M.R.; Lim, H.J.; Young, S.; Cremin, C.; et al. Base excision repair deficiency signatures implicate germline and somatic MUTYH aberrations in pancreatic ductal adeno-carcinoma and breast cancer oncogenesis. Cold Spring Harb. Mol. Case Stud. 2019, 5, doi:/10.1101/mcs.a003681.
- Nones, K.; Johnson, J.; Newell, F.; Patch, A.M.; Thorne, H.; Kazakoff, S.H.; de Luca, X.M.; Parsons, M.T.; Ferguson, K.; Reid, L.E.; et al. Whole-genome sequencing reveals clinically relevant insights into the aetiology of familial breast cancers. Ann. On-col. 2019, 30, 1071–1079, doi:10.1093/annonc/mdz132.
- Viel, A.; Bruselles, A.; Meccia, E.; Fornasarig, M.; Quaia, M.; Canzonieri, V.; Policicchio, E.; Urso, E.D.; Agostini, M.; Genuar-di, M.; et al. A specific mutational signature associated with DNA 8-oxoguanine persistence in MUTYH-defective colorectal cancer. EBioMedicine 2017, 20, 39–49, doi:10.1016/j.ebiom.2017.04.022.
- Tominaga, Y.; Ushijima, Y.; Tsuchimoto, D.; Mishima, M.; Shirakawa, M.; Hirano, S.; Sakumi, K.; Nakabeppu, Y. MUTYH prevents OGG1 or APEX1 from inappropriately processing its substrate or reaction product with its C-terminal domain. Nu-cleic Acids Res. 2004, 32, 3198–3211, doi:10.1093/nar/gkh642.
- Rashid, M.; Fischer, A.; Wilson, C.H.; Tiffen, J.; Rust, A.G.; Stevens, P.; Idziaszczyk, S.; Maynard, J.; Williams, G.T.; Mustonen, V.; et al. Adenoma development in familial adenomatous polyposis and MUTYH-associated polyposis: Somatic landscape and driver genes. J. Pathol. 2016, 238, 98–108, doi:10.1002/path.4643.
- Halazonetis, T.D.; Gorgoulis, V.G.; Bartek, J. An oncogene-induced DNA damage model for cancer development. Science 2008, 319, 1352–1355, doi:10.1126/science.1140735.
- Lefevre, J.H.; Colas, C.; Coulet, F.; Bonilla, C.; Mourra, N.; Flejou, J.F.; Tiret, E.; Bodmer, W.; Soubrier, F.; Parc, Y. MYH bial-lelic mutation can inactivate the two genetic pathways of colorectal cancer by APC or MLH1 transversions. Fam. Cancer 2010, 9, 589–594, doi:10.1007/s10689-010-9367-0.