Bio-hybrid hydrogels consist of a water-swollen hydrophilic polymer network encapsulating or conjugating single biomolecules, or larger and more complex biological constructs like whole cells. By modulating at least one dimension of the hydrogel system at the micro- or nanoscale, the activity of the biological component can be extremely upgraded with clear advantages for the development of therapeutic or diagnostic micro- and nano-devices. Gamma or e-beam irradiation of polymers allow a good control of the chemistry at the micro-/nanoscale with minimal recourse to toxic reactants and solvents. Another potential advantage is to obtain simultaneous sterilization when the absorbed doses are within the sterilization dose range.
- radiation chemistry
- micro-/nano-gel patterns
- nanogels
- e-beam lithography
- nanomedicine
Note: The following contents are extract from your paper. The entry will be online only after author check and submit it.
1.Introduction
Hydrogels have emerged as an important class of functional materials due to their unique structure, tailorable functionalities, and properties, such as high water content, interconnected porosity, softness, and flexibility, that make them resemble biological materials like mucus or the extracellular matrix that surrounds cells, tissues, organs, or entire organisms [1,2][1][2]. They can be classified into physical and chemical or permanent gels, depending on the nature of the crosslinking points [3]. Molecular entanglements and secondary forces are responsible for the crosslinks in physical gels. This makes them “reversible”, meaning that they can dissolve upon a change of the environmental conditions or just by prolonged exposure to aqueous solutions. Contrariwise, permanent or chemical gels are networks with covalent bonds as crosslinking points.
Hydrogels can be produced either from polymerization and simultaneous crosslinking of hydrophilic monomers in the presence of polyfunctional crosslinking agents, or directly by crosslinking of hydrophilic polymers. Residual amounts of monomers, initiators, catalysts, and their side products can often result in undesirable characteristics, such as color, chemical reactivity, and potential toxicity. Purification is typically performed by extraction into excess water and may take up to several weeks to be completed. Therefore, the demand for facile and safer synthetic approaches makes single component-based processes particularly attractive.
Irradiation with either gamma rays or accelerated electron beams has been successfully applied to produce hydrogels from water-soluble, biocompatible synthetic polymers, such as poly(acrylic acid) poly(vinyl alcohol), poly(vinylpyrrolidone), poly(ethylene glycol) and polyacrylamide, [4][5] [4,5] polysaccharides [6,7,8,9][6][7][8][9], and polyaminoacids [10,11][10][11]. One distinct advantage of the recourse to high energy irradiation is that sterilization can also be simultaneously achieved, if suitable doses are imparted. Alternative, reagent-free approaches rely on UV-irradiation of directly photo-crosslinkable polymeric systems, autoclave, or microwave radiation thermal treatments. Direct photo-crosslinking requires the presence of photoactive groups in the polymer, such as coumarin, cinnamic acid, anthracene, or dimethylmaleimide [12,13][12][13]. Not many biocompatible water-soluble polymers possess these functional groups. Therefore, it often requires prior ad hoc polymer functionalization. Autoclave or microwave-assisted thermal cross-linking methods are inexpensive and safe, but they do not allow easy control of the locus of reaction, therefore they are not suitable for producing micro-/nanopatterned hydrogel surfaces [14] [14].
One further important step towards the obtainment of hydrogels that can accomplish specific roles in biosensing, therapy, or regenerative medicine is the combination of these substrates with biological molecules, such as peptides, antibodies or their portions, oligonucleotides, enzymes, hormones, and several other therapeutic proteins. The incorporated or grafted biological molecules can grant specific functionality [15], while the hydrogel as substrate can provide the biological molecules with an aqueous, three-dimensional microenvironment that can help preserve their structure and functionality [16,17][16][17].
2. Radiation Chemistry and Physics in Hydrogel Technology
Absorption of ionizing radiation such as γ-photons and high energy electrons induces ionization and excitation of matter which subsequently leads to the formation of reactive radicals [18]. The further reactions of the radicals depend on the structure and viscosity of the material and also on the amount of energy deposited in the material. The latter is referred to as the absorbed dose [18].
When irradiating a pure polymer, absorption of the radiation energy will lead to the formation of radicals [19]. The radicals formed on the polymer chain can react with radical sites on other polymer chains or on the same polymer chain. These radical-radical reactions can either lead to the formation of covalent bonds, i.e., crosslinking, or to disproportionation where one radical site oxidizes the other radical site. The competition between crosslinking and disproportionation depends on the structure of the radicals involved and cannot be affected by external factors [20]. In addition, the radicals formed can also undergo unimolecular scission (fragmentation) reactions. The competition between the unimolecular scission reaction and the bimolecular radical-radical reaction depends on the structure of the radicals as well as on the concentration of radical sites in the system. The latter is directly influenced by the radiation energy deposited per unit time, also known as the dose rate. In systems exposed to air, the polymer radicals formed may also react with molecular oxygen which will enable additional reactions where chain scission and incorporation of oxidized functional groups are two probable outcomes. The possible reactions in an irradiated polymer system are schematically illustrated in Figure 1 [19,20].
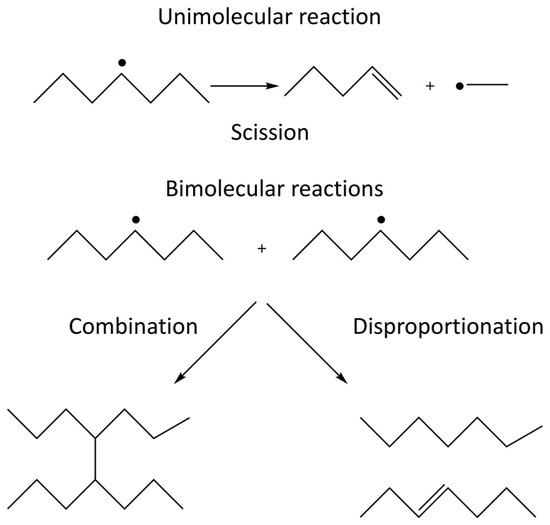
Figure 1. Unimolecular and bimolecular reactions occurring in irradiated polymers. The unimolecular reaction is scission (depolymerization) and the bimolecular reactions are radical-radical combination and disproportionation.
In this paper we primarily focus on crosslinking of hydrophilic polymer chains in order to produce polymer networks that, upon exposure to water, undergo swelling to produce hydrogels. Chain scission and functionalization occur as concurrent reactions.
In systems composed of more than one component, the absorbed radiation energy is distributed between the components according to their relative masses. In aqueous solutions, most of the radiation energy is absorbed by the water. The aqueous radiolysis products are HO•, H•, eaq−, H2O2, H2, and H3O+ [18]. HO• and H• are capable of abstracting H-atoms from most polymers and thereby produce the same types of radicals as the ones that are formed upon irradiation of the pure polymer. However, radicals formed as a consequence of direct scission of the backbone, due to direct absorption of the radiation energy by the polymer, will occur to a much lesser extent in solution. In dilute aqueous polymer solutions, the polymer radical formation can be almost exclusively attributed to the absorption of ionizing radiation by the water and is therefore referred to as an “indirect effect”, while the polymer radical formation in the irradiated pure polymer is referred to as a “direct effect”.
The polymer radicals formed in aqueous solution can react in the same way as the radicals formed in the pure polymer system. However, the viscosity of the systems as well as the polymer concentration and thereby the polymer radical concentration and spatial distribution differ significantly [18]. This has a major impact on the kinetics of the reactions both in absolute and relative terms. At a sufficiently high polymer concentration, the radical-radical crosslinking reactions will produce a polymer network in the solution, hence a hydrogel [4].
The radiation chemical yields (i.e., the amount of produced or consumed species per absorbed unit of radiation energy) of the aqueous radiolysis products are well known. This enables thorough design of the syntheses in aqueous solution. In general, the radiation chemical yields depend on the type of irradiation and on the radiation energy. However, high energy electrons and γ-photons have the same radiation chemical yields. The radiation chemical yields of the aqueous radiolysis products upon exposure to γ-radiation or high energy electrons are the following (in μmol J−1): HO• (0.28), H• (0.062), eaq− (0.28), H2O2 (0.073), H2 (0.047), and H3O+ (0.28) [18]. Interestingly, N2O dissolved in water can convert the solvated electron (eaq−) to a hydroxyl radical and thereby double the radiation chemical yield. For this reason, N2O saturation is often employed when performing radiation synthesis of aqueous systems (provided that the hydroxyl radical is the desired reactant).
While γ-photons are in general very penetrating, electrons have a limited penetration depth. The penetration depth of electrons in a given material is strongly dependent on the electron energy where a higher electron energy corresponds to a larger penetration depth [18]. The direct implication of this is that modification of thicker materials requires higher electron energies. For this reason, radiation processing of thin films can be performed with keV electrons, while processing larger volumes of solutions requires MeV electrons. As a consequence, completely different irradiation facilities must be used.
The transfer of energy from the incident high energy electron to the absorbing material can be quantified in terms of linear energy transfer (LET), which is a measure of absorbed energy per unit path length. The LET value in a given absorber depends on the type of radiation and the energy. In general, the LET value increases with decreasing energy. Therefore, low energy electrons will have a higher LET than high energy electrons. The direct consequence of this is that the distance between ionizations (and thereby radical formation) will be shorter for low energy electrons than for high energy electrons. In other words the radical concentration will be higher and thereby also the probability for radical-radical reactions.
3. Radiation Engineering of Hydrogels at the Micro-/Nanoscale
3.1. Micropatterned Hydrogels by Electron Beam Lithography
Accelerated electron-beams can be used to create hydrogel patterns at the nano- and micro-length scales on surfaces, with a typical top-down approach. The methodology, known as electron beam lithography (EBL), is a well-established precision tool for nanoscale pattern generation. The polymers are deposited in the form of thin dry films (e-beam resists) on the pre-treated (polished, degreased, etched, and coated with a primer) substrates and subjected to localized irradiation with focused accelerated electron beams of relatively low energy (in the keV range). The pattern is then developed by rinsing the surface with water. This will wash away polymer chains that are not crosslinked or grafted to the substrate. Conversely, crosslinked polymer chains that are also grafted to the substrate will remain on the surface and the network will take up water to yield a hydrogel. EBL has the advantage of being able to generate surface-patterned structures with arbitrary shapes and feature size down to a few tens of nanometers [21,22][21][22].
The first microscopic hydrogel patterns were created using spatially resolved electron beams from scanning electron microscopes (SEM) and solvent-free polyethylene glycol (PEG or PEO) films as e-beam resist [23,24][23][24]. Several further reports describe the fabrication of micro-/nano-gel patterns by EBL using other polymers, such as poly (vinyl methyl ether) [25], poly (vinyl pyrrolidone) (PVP) [26], polyamidoamine [27], oligo(ethylene glycol) methacrylate [28], and star PEGs with functional end groups [29].
It is generally proposed that irradiation of these polymers in vacuum form carbon-centered radicals as a result of the direct effects of irradiation, that can then be involved in several chain-chain and chain-substrate radical reactions. As stated above, chain scission and crosslinking occur simultaneously during electron irradiation, but at different rates depending on the conditions. The overall response of the e-beam resist is thus determined by the dominant of the two competing processes. During the course of irradiation, increased crosslinking will increase the viscosity of the film and thereby reduce the rate constant for bimolecular reactions. Hence, the competition between the processes will change. Conditions to achieve critical crosslinking (i.e., minimum one crosslinking point per chain) and grafting to the underlying surface must be simultaneously fulfilled in order to form stable surface patterned micro-/nanogels.
When discussing more in detail how it is possible to control the locus of radiation chemical changes of an e-beam resist, it should be recalled that when an accelerated electron impinges on a solid target from vacuum, it undergoes both elastic and inelastic scattering. The nature and extent of these two processes depend on the nature of the absorbent material and initial energy of the incident electron. Electron scattering will have a direct effect on the spatial resolution of EBL. In Figure 2a, the results of a Monte Carlo simulation describing the trajectory of 1000 electrons with energies of 2, 5, 10, or 30 keV across a 100 nm thick PEG resist on a Si substrate are represented [30].
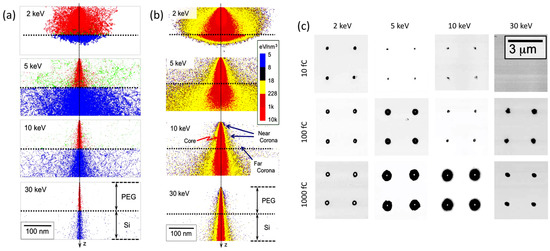
Figure 2. (a) Monte Carlo simulation describing the trajectory of 1000 electrons with energies of 2, 5, 10, or 30 keV across a 100 nm thick solvent-free polyethylene glycol (PEG) resist on a Si substrate; primary electrons of the beam are represented in red, scattered electrons in blue, backscattered electrons in green. (b) Distribution of deposited energy (c). Phase-contrast atomic force microscopy (AFM) images of PEG microgels patterned on silanized Si surfaces with different incident electron energies and different point doses. Based on [30].
Primary electrons (red) that pass through the resist into the underlaying Si layer are scattered to higher angles (blue). Backscattered electrons (green) originate from both media and when generated on Si they can re-emerge into the resist at a large distance from the primary electrons they originated from. This effect, called “proximity effect”, affects the precision of EBL. Indeed, the distribution of deposited energy (i.e., dose distribution) at and around the point of electron-beam incidence can be very non-uniform (see Figure 2b). In high dose regions, called “core” (red, in Figure 2b), crosslinking can be so extensive that the polymer becomes very rigid and not quite gel-like. In the surrounding area, (yellow, in Figure 2b), the absorbed doses are lower and can mainly be attributed to primary electrons scattered from their incident direction (Z) at the top (“near-corona”), and to backscattered electrons at the bottom (“far-corona”). The density of crosslinks of the near-corona is lower and the material upon development can become a soft, hydrated gel. The far-corona has similar gel-like properties to the near-corona but is localized at the interface with the substrate and can extend many tens of nanometers and more away from the core. For low energy electrons, the near-corona can be thick and there can essentially be no far-corona. The reason is that the number of backscattered electrons is low and they have insufficient energy to travel far from the core. In contrast, higher energy electrons can be backscattered from the substrate and can again induce chemical reactions on their second trip through the resist. Yet, the reactions caused by these electrons may not be sufficiently extensive to graft all polymer chains to the network and all network portions to the substrate. Consequently, isolated portions of networks that have formed happen to be removed during development, generating discontinuous layers of gels even at long distance from the core. In some cases, only grafted single chains can be found. In conclusion, the structure and extent of the far-corona is strongly affected by the irradiation conditions.
Figure 2c shows phase-contrast atomic force microscopy (AFM) images of PEG microgels patterned with different incident electron energies and different exposures. The dark spots correspond to microgels. The white spots in the center of some microgels indicate a difference in mechanical properties of the core with respect to the shell. The different parts of the structure, depending on the degree and density of crosslinking, can have a very different affinity to molecules, biomolecules, and cells. PEG is generally used for its non-adhesive, antifouling properties that minimize non-specific protein adsorption [31], trusting on specific functional groups in PEG chain ends and their reaction with biomolecules for the specific interaction or reaction with the complementary dye, protein, or membrane cell receptor. The hard core might not be hydrated enough to present these “antifouling” properties, but depending on the size of the PEG segments stretching out in the solvent from the loosely crosslinked corona, the core can also be shielded by protein absorption. On the other hand, even a monolayer of PEG is sufficient, for example, to convert a surface from protein-adhesive to protein non-adhesive [30]. Hence, the choice of the electron-exposure conditions for patterning the resist can control the subsequent bio-interactive properties of the micro-/nano-patterned hydrogels over length scales that are relevant to both proteins and cells.
3.2. Radiation Engineering of Hydrogel Nanoparticles
As described above, hydrogels are formed when aqueous polymer solutions are irradiated with ionizing radiation. Under irradiation conditions where the dose rate is high enough to ensure the coexistence of multiple radical sites per polymer chain, the radical-radical reactions can be intramolecular as well as intermolecular. While the intermolecular crosslinking builds a more macroscopic hydrogel, the intramolecular crosslinking can produce hydrogel nanoparticles (nanogels) where the smallest entity is a single polymer chain that has been intramolecularly crosslinked [32] [32].
The impact of polymer concentration and dose rate on nanogel size have been thoroughly investigated [4,5,33,34,35,36][33][34][35][36]. Intramolecular radical-radical reactions, and thereby also nanogel formation, is favored by low polymer concentration (below the critical chain overlap concentration of the polymer) and high dose rate. High dose rate will promote formation of multiple radical sites per polymer chain while the low polymer concentration will suppress intermolecular interactions in general and thereby also intermolecular radical-radical reactions. When changing the polymer concentration from high to low, the conditions change from favoring formation of macrogels to single chain nanogels. At concentrations below but still relatively close to the critical chain overlap concentration, intermolecular radical-radical crosslinking can initially occur, leading to a decrease in particle concentration after which intramolecular crosslinking becomes dominating. Hence, particle size no longer significantly increases. Extensive intramolecular crosslinking restrains the polymer chain segment mobility and thereby reduces the possibility of radicals to get in close enough proximity to react. This, in turn, progressively reduces the rate of intramolecular radical-radical combination. [33,35][33][35] In Figure 3, the reactions involving polymer radicals in aqueous solution are schematically described.
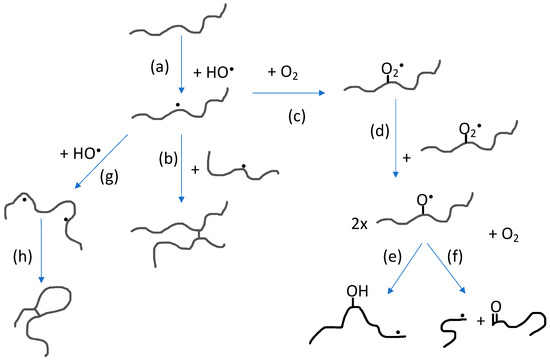
Figure 3. Reactions involving polymer radicals in aqueous solution: (a) Hydrogen abstraction by hydroxyl radicals producing polymer radicals; (b) intermolecular radical combination (the competing disproportionation is not shown); (c) reaction with molecular oxygen; (d) reaction between peroxyl radicals; (e) intramolecular H-transfer; (f) oxyl radical β-scission; (g) hydrogen abstraction from polymer radical producing multiple radical sites; and (h) intramolecular radical combination (also in this case, disproportionation is not shown).
A particular mode of irradiation that has been used in the synthesis of nanogels is the use of pulsed electron beams from electron accelerators. The dose rate during the pulse is usually extremely high but between the pulses there is a relatively long period during which the solution is not irradiated [33,37][37]. Under these conditions it is more relevant to discuss the dose per pulse rather than the average dose rate.
As previously mentioned, radical-radical reactions lead to crosslinking as well as disproportionation. The latter reaction does not change the polymer chain length but it introduces alkene functionality on one of the two reacting radical sites according to the general reaction shown in Figure 1. The alkene functionality can be employed in further functionalization of the nanogel.
As mentioned above, by saturating an aqueous polymer solution with N2O, the hydroxyl radical yield is doubled. For N2O-saturated, dilute polymer solutions, the scavenging of hydroxyl radicals by the polymer is not complete and OH-radical recombination competes with polymer radical formation. This leads to the production of H2O2 and subsequently also to the formation of molecular oxygen [35,37]. Molecular oxygen reacts rapidly with C-centered radicals, a reaction that will compete with radical-radical reactions and result in oxidation of the polymer. When using PVP as starting material for the nanogel synthesis, it has been shown that primary amino groups as well as carboxyl groups are formed during the irradiation and the number of groups per particle increases with increasing dose [33]. This is a clear advantage when using the nanogels as theranostic nanocarriers in medicine.
References
- Calò, E.; Khutoryanskiy, V.V. Biomedical applications of hydrogels: A review of patents and commercial products. Eur. Polym. J. 2015 65, 252–267, doi:10.1016/j.eurpolymj.2014.11.024.
- Lieleg, O.; Ribbeck, K. Biological hydrogels as selective diffusion barriers. Trends Cell Biol. 2011, 21, 543–551, doi:10.1016/j.tcb.2011.06.002.
- Hoffman, A.S. Hydrogels for biomedical appilcations. Adv. Drug Deliv. Rev. 2002, 54, 3–12, doi:10.1016/s0169-409x(01)00239-3.
- Rosiak, J.M.; Ulański, P. Synthesis of hydrogels by irradiation of polymers in aqueous solution. Radiat. Phys. Chem. 1999, 55, 139–151, doi:10.1016/S0969-806X(98)00319-3.
- Sabatino, M.A.; Bulone, D.; Veres, M.; Spinella, A.; Spadaro, G.; Dispenza, C. Structure of e-beam sculptured poly(N-vinylpyrrolidone) networks across different length-scales, from macro to nano. Polymer 2013, 54, 54–64, doi:10.1016/j.polymer.2012.11.031.
- Wach, R.A.; Mitomo, H.; Yoshii, F.; Kume, T. Hydrogel of radiation-induced cross-linked hydroxypropylcellulose. Macromol. Mater. Eng. 2002, 287, 285–295, doi:10.1002/1439-2054(20020401)287:4%3C285::AID-MAME285%3E3.0.CO;2-3.
- Abdel Ghaffar, A.M.; Radwan, RR.; Ali, H.E. Radiation synthesis of poly(starch/acrylic acid) pH sensitive hydrogel for rutin controlled release. Int. J. Biol. Macromol. 2016, 92, 957–964, doi:10.1016/j.ijbiomac.2016.07.079.
- Zhao, L.; Gwon, H.-J.; Lim, Y.-M.; Nho, Y.C.; Kim, S.Y. Hyaluronic acid/chondroitin sulfate-based hydrogel prepared by gamma irradiation technique. Carbohydr. Polym. 2014, 102, 598–605, doi:10.1016/j.carbpol.2013.11.048.
- Wach, R.A.; Adamus-Wlodarczyk, A.; Olejnik, A.K.; Matusiak, M.; Tranquilan-Aranilla, C.; Ulanski, P. Carboxymethylchi-tosan hydrogel manufactured by radiation-induced crosslinking as potential nerve regeneration guide scaffold. React. Funct. Polym. 2020, 152, 104588, doi:10.1016/j.reactfunctpolym.2020.104588.
- Spadaro, G.; Dispenza, C.; Giammona, G.; Pitarresi, G.; Cavallaro, G. Cytarabine release from b,a-poly(N-hydroxyethyl)-DL-aspartamide matrices cross-linked through gamma-radiation. Biomaterials 1996, 17, 953–958, doi:10.1016/0142-9612(96)84668-0.
- Visotzki, E.I.; Hennes, M.; Schuldt, C.; Engert, F.; Knolle, W.; Decker, U.; Kas, J.A.; Zink, M.; Mayr, S.G. Tayloring the mate-rial properties of gelatin hydrogels by high energy electron radiation. J. Mater. Chem. B 2014, 2, 4297–4309, doi:10.1039/C4TB00429A.
- Ravve, A. Photocrosslinkable polymers. In Light-Associated Reactions of Synthetic Polymers; Ravve, A., Ed., Springer, New York, NY, USA, 2006; pp. 201–244, doi:10.1007/0-387-36414-5_4.
- Shirai, M. Photocrosslinkable polymers with degradable properties. Polym. J. 2014, 46, 859–865, doi:10.1038/pj.2014.79.
- Cook, J.P.; Goodall, G.W.; Khutoryanskaya, O.V.; Khutoryanskiy, V.V. Microwave-Assisted Hydrogel Synthesis: A New Method for Crosslinking Polymers in Aqueous Solutions. Macromol. Rapid Commun. 2012, 33, 332–336, doi:10.1002/marc.201100742.
- Zhang, D.; Xu, X.; long, X.; Cheng, K.; Li, J. Advances in biomolecules inspired polymeric material decorated interfaces for biological applications. Biomater. Sci. 2019, 7, 3984–3999, doi:10.1039/C9BM00746F.
- Freudenberg, U.; Liang, Y.; Kiick, K.L.; Werner, C. Glycosaminoglycan-Based Biohybrid Hydrogels: A Sweet and Smart Choice for Multifunctional Biomaterials. Adv. Mater. 2016, 28, 8861–8891, doi:10.1002/adma.201601908.
- Roshanbinfar, K.; Vogt, L.; Greber, B.; Diecke, S.; Boccaccini, A.R.; Scheibel, T.; Engel, F.B. Electroconductive biohybrid hy-drogel for enhanced maturation and beating properties of engineered cardiac tissues. Adv. Funct. Mater. 2018, 28, 1803951, doi:10.1002/adfm.201803951.
- Spinks, J.W.T.; Woods, R.J. An Introduction to Radiation Chemistry, 3rd ed.; John-Wiley and Sons, Inc.: New York, TO, Canada, 1990; doi:10.1002/bbpc.19910950346.
- Charlesby, A. Irradiation Effects on Polymers; Clegg, D.W., Collyer, A.A., Eds.; Elsevier Applied Science: New York, NY, USA, 1991.
- Alfassi, Z.B. General Aspects of the Chemistry of Radicals; Wiley: New York, NY, USA, 1999.
- Abd El-Rehim, H.A.; Hegazy, E.S.A.; Hamed, A.A.; Swilem, A.E. Controlling the size and swellability of stimuli-responsive polyvinylpyrrolidone-poly(acrylic acid) nanogels synthesized by gamma radiation-induced template polymerization. Eur. Polym.J. 2013, 49, 601–612, doi:10.1016/j.eurpolymj.2012.12.002.
- Kolodziej, C.M.; Maynard, H.D. Electron-beam lithography for patterning biomolecules at the micron and nanometer scale Chem. Mater. 2012, 24, 774–780, doi:10.1021/cm202669f2.
- Abd El-Rehim, H.A.; Swilem, A.E.; Klingner, A.; Hegazy, E.S.A.; Hamed, A.A. Developing the potential ophthalmic applications of pilocarpine entrapped into polyvinylpyrrolidone-poly(acrylic acid) nanogel dispersions prepared by γ radiation. Biomacromolecules 2013, 14, 688–698. doi:10.1021/bm301742m.
- Krsko, P.; Sukhishvili, S.; Mansfield, M.; Clancy, R.; Libera, M. Electron-beam surface-patterned poly(ethylene glycol) micro-hydrogels Langmuir 2003, 19, 5618–5625, doi:10.1021/la034157r.
- Schmidt, T.; Mönch, J.I.; Arndt, K.-F. Temperature-sensitive hydrogel pattern by electron-beam lithography. Macromol. Mater. Eng. 2006, 291, 755–761, doi:10.1002/mame.200600057.
- Burkert, S.; Schmidt, T.; Gohs, U.; Mönch, I.; Arndt, K.-F. Patterning of thin poly(N-vinyl pyrrolidone) films on silicon sub-strates by electron beam lithography. J. Appl. Polym. Sci. 2007, 106, 534–539, doi:10.1002/app.26592.
- Dos Reis, G.; Fenili, F.; Gianfelice, A.; Bongiorno, G.; Marchesi, D.; Scopelliti, P.E.; Borgonovo, A.; Podestà, A.; Indrieri, M.; Ranucci, E.; et al. Direct microfabrication of topographical and chemical cues for the guided growth of neural cell networks on polyamidoamine hydrogels. Macromol. Biosci. 2010, 10, 842–852, doi:10.1002/mabi.200900410.
- Bat, E.; Lin, E.W.; Saxer, S.; Maynard, H.D. Morphing hydrogel patterns by thermo-reversible fluorescence switching. Macromol. Rapid Comm. 2014, 35, 1260–1265, doi:10.1002/marc.201400160.
- Mancini, R.J.; Paluck, S.J.; Bat, E.; Maynard, H.D. Encapsulated hydrogels by E-beam lithography and their use in enzyme cascade reactions. Langmuir 2016, 32, 4043–4051, doi:10.1021/acs.langmuir.6b00560.
- Wang, Y.; Firlar, E.; Dai, X.; Libera, M. Poly (ethylene glycol) as a biointeractive electron-beam resist. J. Polym. Sci. Part B Polym. Phys. 2013, 51, 1543–1554, doi:10.1002/polb.23367.
- Lowe, S.; O’Brien-Simpson, N.M.; Luke, A.; Connal, L.A. Antibiofouling polymer interfaces: Poly (ethylene glycol) and other promising candidates. Polym. Chem. 2015, 6, 198–212, doi:10.1039/c4py01356e.
- Ulanski, P.; Ianik, I.; Rosiak, J.M. Radiation formation of polymeric nanogels. Radiat. Phys. Chem. 1998, 52, 289–294.
- Dispenza, C.; Sabatino, M.A.; Grimaldi, N.; Mangione, M.R.; Walo, M.; Murugan, E.; Jonsson, M. On the origin of function-alization in one-pot radiation synthesis of nanogels from aqueous polymer solutions. Rsc Adv. 2016, 6, 2582–2591, doi:10.1039/c5ra23926e.
- Dispenza, C.; Spadaro, G.; Jonsson, M. Radiation engineering of multifunctional nanogels. Top. Curr. Chem. 2016, 374, 69, doi:10.1007/s41061-016-0071-x.
- Ditta, L.A.; Dahlgren, B.; Sabatino, M.A.; Dispenza, C.; Jonsson, M. The role of molecular oxygen in the formation of radia-tion-engineered multifunctional nanogels. Eur. Polym. J. 2019, 114, 164–175, doi:10.1016/j.eurpolymj.2019.02.020.
- Dispenza, C.; Sabatino, M.A.; Grimaldi, N.; Dahlgren, B.; Al-Sheikhly, M.; Wishart, J.F.; Tsinas, Z.; Poster, D.L.; Jonsson, M. On the nature of macroradicals formed upon radiolysis of aqueous poly(N-vinylpyrrolidone solutions. Radiat. Phys. Chem. 2020, 174, 108900, doi:10.1016/j.radphyschem.2020.108900.
- Dahlgren, B.; Sabatino, M.A.; Dispenza, C.; Jonsson, M. Numerical simulations of nanogel synthesis using pulsed electron beam. Macromol. Theory Simul. 2020, 29, 1900046, doi:10.1002/mats.201900046.