The cold-shock domain has a deceptively simple architecture but supports a complex biology. Cold-shock domains in human proteins are often associated with natively unfolded protein segments and more rarely with other folded domains. Human proteins containing cold-shock domains bind single-stranded DNA and/or RNA and serve a large variety of roles in regulating transcription, DNA-damage repair, RNA splicing, translation, stability and sequestration.
- cold-shock domain
- cold-shock protein
1. Introduction
Proteins are made of compact domains with defined three-dimensional folding and of natively unstructured polypeptide segments. These globular domains are recurrent structural elements, serving as parts sets of molecular evolution and often appearing in multiple proteins that may or may not share common biochemical or biological functions. The number of domain folds is limited; an early hypothesis speculated about the presence of “one thousand families for the molecular biologist” [1]. As with domain folds in the entire protein universe, there is a limited repertoire of RNA-binding domains (RBDs) [2] including the cold-shock domain (CSD). Canonical RBDs preferentially bind short single-stranded sequence motifs in RNA, but binding to structured regions of RNA is also observed [3].
The total number of human RNA-binding proteins (RBPs) is estimated at more than 800 in both proliferating HeLa cells [4] and in an embryonic kidney cell line [5]. An analysis of the RNA interactome to the sub-domain level revealed more than 1100 RNA-binding sites in more than 500 RBPs including CSD-containing proteins [6].
The total number of human RNA-binding proteins (RBPs) is estimated at more than 800 in both proliferating HeLa cells [4] and in an embryonic kidney cell line [5]. An analysis of the RNA interactome to the sub-domain level revealed more than 1100 RNA-binding sites in more than 500 RBPs including CSD-containing proteins [6].
2. Definition and Basic Properties of Cold-Shock Domains
Almost three decades ago, a common oligonucleotide/oligosaccharide-binding (OB) fold was identified in four proteins unrelated by sequence: staphylococcal nuclease, the anticodon-binding domain of aspartyl-tRNA synthetase, and the β-subunits of two
Escherichia coli toxins, heat-labile enterotoxin and verotoxin-1. The OB fold as present in these proteins is characterized by 70–150 amino-acid (aa) residues organized into a five-stranded all-antiparallel β-barrel which is frequently capped by an α-helix located between strands β3 and β4. Oligonucleotide and oligosaccharide ligands bind to a conserved surface area on the barrel [7]. There is little to no sequence conservation between OB-fold proteins, but ssDNA and ssRNA bind with conserved polarity to oligonucleotide-binding family members [8].
toxins, heat-labile enterotoxin and verotoxin-1. The OB fold as present in these proteins is characterized by 70–150 amino-acid (aa) residues organized into a five-stranded all-antiparallel β-barrel which is frequently capped by an α-helix located between strands β3 and β4. Oligonucleotide and oligosaccharide ligands bind to a conserved surface area on the barrel [15]. There is little to no sequence conservation between OB-fold proteins, but ssDNA and ssRNA bind with conserved polarity to oligonucleotide-binding family members [16].
OB-fold proteins including staphylococcal nuclease, the bacterial enterotoxins and the MOP (molybdate/tungstate binding)- and TIMP (tissue inhibitor of metalloproteases)-like proteins have been classified into superfamilies. The nucleic acid-binding proteins constitute the largest OB-fold superfamily, including protein families such as the single-strand DNA-binding (SSB) proteins and RNA-binding RNB domain-like proteins. Among the nucleic acid-binding proteins, the cold-shock DNA-binding domain-like proteins constitute the largest family. This protein family can be further divided into proteins containing (proper) cold-shock domains, which are the subject of this review, and other domains including the S1, IF2-type S1, S12, S17, and S28e domains first identified in proteins of the small ribosomal subunit [9]. The taxonomic distribution of the abundant S1 domain-containing family of OB-fold proteins was recently reviewed [10]. Frequently, multiple OB domains are present in oligomeric proteins or within one polypeptide chain. The human CTC1-STN1-TEN1 (CST) complex essential for telomere maintenance, may serve as an impressive example. Here, all subunits contain OB-fold domains two of which (OB-F and OB-G of CTC1) are involved in ssDNA binding as demonstrated by cryo-electron microscopy (cryo-EM) [11].
OB-fold proteins including staphylococcal nuclease, the bacterial enterotoxins and the MOP (molybdate/tungstate binding)- and TIMP (tissue inhibitor of metalloproteases)-like proteins have been classified into superfamilies. The nucleic acid-binding proteins constitute the largest OB-fold superfamily, including protein families such as the single-strand DNA-binding (SSB) proteins and RNA-binding RNB domain-like proteins. Among the nucleic acid-binding proteins, the cold-shock DNA-binding domain-like proteins constitute the largest family. This protein family can be further divided into proteins containing (proper) cold-shock domains, which are the subject of this review, and other domains including the S1, IF2-type S1, S12, S17, and S28e domains first identified in proteins of the small ribosomal subunit [17]. The taxonomic distribution of the abundant S1 domain-containing family of OB-fold proteins was recently reviewed [18]. Frequently, multiple OB domains are present in oligomeric proteins or within one polypeptide chain. The human CTC1-STN1-TEN1 (CST) complex essential for telomere maintenance, may serve as an impressive example. Here, all subunits contain OB-fold domains two of which (OB-F and OB-G of CTC1) are involved in ssDNA binding as demonstrated by cryo-electron microscopy (cryo-EM) [19].
In contrast to the wider group of OB-fold proteins, the bacterial CSPs and eukaryotic CSDs are of very similar length of about 70 aa and share clearly conserved sequences. The presence of RNP1 and RNP2 sequence motifs [12] provided an early hint towards a function of CSDs in binding single-stranded nucleic acids. As in other RBPs, the RNP motifs [13] are placed within β-strands. These RNP motifs contain some of the most highly conserved residues in the set of representative bacterial CSPs and eukaryotic CSDs displayed in
In contrast to the wider group of OB-fold proteins, the bacterial CSPs and eukaryotic CSDs are of very similar length of about 70 aa and share clearly conserved sequences. The presence of RNP1 and RNP2 sequence motifs [20] provided an early hint towards a function of CSDs in binding single-stranded nucleic acids. As in other RBPs, the RNP motifs [21] are placed within β-strands. These RNP motifs contain some of the most highly conserved residues in the set of representative bacterial CSPs and eukaryotic CSDs displayed in
. In general, sequence conservation in CSPs and CSDs is higher in β-strands than in loop regions. Whereas bacterial CSPs are small proteins consisting, as a rule, of a single CSD only, their eukaryotic homologs are proteins of variable length and domain composition.

Figure 1.
Sequence alignment of representative bacterial CSPs and CSDs from human proteins. The
Xenopus laevis FRGY1 and FRGY2 (YBOX1, YBX2A, YBX2B) proteins are also included. Human CSDE1 contains five CSDs, all other proteins contain or consist of a single CSD. Proteins are identified by their Uniprot [14] entry number and name. The secondary-structure annotation atop the sequence follows
FRGY1 and FRGY2 (YBOX1, YBX2A, YBX2B) proteins are also included. Human CSDE1 contains five CSDs, all other proteins contain or consist of a single CSD. Proteins are identified by their Uniprot [22] entry number and name. The secondary-structure annotation atop the sequence follows
BsCspB, the first CSP for which a crystal structure was determined [14]. Residues conserved across all aligned CSDs are highlighted on dark blue background and shown with capital letters in the consensus sequence. Residues conserved in ≥50% of the sequences are shown on a light blue background and with lower-case letters in the consensus. Sequences were aligned using the Clustal Omega server [15]. The sequence motifs RNP1 ([YF]-G-F-I) and RNP2 ([YF]-[YF]-H) are associated with RNA binding and indicated according to Prosite [16].
CspB, the first CSP for which a crystal structure was determined [23]. Residues conserved across all aligned CSDs are highlighted on dark blue background and shown with capital letters in the consensus sequence. Residues conserved in ≥50% of the sequences are shown on a light blue background and with lower-case letters in the consensus. Sequences were aligned using the Clustal Omega server [24]. The sequence motifs RNP1 ([YF]-G-F-I) and RNP2 ([YF]-[YF]-H) are associated with RNA binding and indicated according to Prosite [25].
3. Structure of Cold-Shock Domains
The CSD is a simplified version of the OB fold lacking the α-helix.
B. subtilis CspB was the first CSP to be crystallized [17], and its structure, determined from two crystal forms, set the paradigm for bacterial CSP and eukaryotic CSD conformation. The polypeptide chain is organized into an antiparallel five-stranded β-barrel with connecting loops of variable length. In the β-barrel, a three-stranded β-sheet and a two-stranded β-ladder are recognizable which are linked by only a few backbone hydrogen bonds. In spite of its small size of ~70 aa, the CSD contains a fully formed hydrophobic core. The presence of exposed aromatic residues on a basic protein surface strongly suggested a role in binding single-stranded nucleic acids, and ssDNA binding was confirmed in a gel-shift experiment [18]. The core findings of the crystallographic analysis of
CspB was the first CSP to be crystallized [93], and its structure, determined from two crystal forms, set the paradigm for bacterial CSP and eukaryotic CSD conformation. The polypeptide chain is organized into an antiparallel five-stranded β-barrel with connecting loops of variable length. In the β-barrel, a three-stranded β-sheet and a two-stranded β-ladder are recognizable which are linked by only a few backbone hydrogen bonds. In spite of its small size of ~70 aa, the CSD contains a fully formed hydrophobic core. The presence of exposed aromatic residues on a basic protein surface strongly suggested a role in binding single-stranded nucleic acids, and ssDNA binding was confirmed in a gel-shift experiment [23]. The core findings of the crystallographic analysis of
BsCspB were essentially confirmed by the solution structure of the protein determined by nuclear magnetic resonance (NMR) spectroscopy [19]. Subsequently, a crystal structure of
CspB were essentially confirmed by the solution structure of the protein determined by nuclear magnetic resonance (NMR) spectroscopy [94]. Subsequently, a crystal structure of
Ec
CspA was determined at 2-Å resolution which revealed the same architecture as that of
BsCspB and a conserved nucleic acid-binding surface [20]. The solution NMR structure of
CspB and a conserved nucleic acid-binding surface [95]. The solution NMR structure of
EcCspA was in general agreement with the crystallographic analysis and identified nine aromatic and two basic residues in binding to a 24-nucleotide ssDNA [21].
CspA was in general agreement with the crystallographic analysis and identified nine aromatic and two basic residues in binding to a 24-nucleotide ssDNA [96].
The crystal structure of
Bc
CspB at atomic resolution of 1.17 Å (
Figure 2a,b) confirms the expected close conformational similarity with
3a,b) confirms the expected close conformational similarity with
Bs
CspB and suggests that the surface charge distribution (
Figure 2c) may be linked to the enhanced thermal stability of this protein from the thermophilic
3c) may be linked to the enhanced thermal stability of this protein from the thermophilic
Bacillus caldolyticus [22]. The NMR structure of the homologous
[97]. The NMR structure of the homologous
Tm
Csp from the hyperthermophilic
Thermotoga maritima suggests residues mediating enhanced thermal stability in this CSP [23]. The structure of a psychrophilic CSP from
suggests residues mediating enhanced thermal stability in this CSP [98]. The structure of a psychrophilic CSP from
Listeria monocytogenes
determined by NMR resembles other bacterial CSP structures, but with a melting transition at 40 °C
LmCspA has reduced thermostability [24]. A variation in the structures of bacterial CSPs is offered by the NMR analysis of the single CSP from
CspA has reduced thermostability [99]. A variation in the structures of bacterial CSPs is offered by the NMR analysis of the single CSP from
Rickettsia rickettsii, which shows a canonical CSP structure with the insertion of a short segment of α-helix in the long loop L3 [25].
, which shows a canonical CSP structure with the insertion of a short segment of α-helix in the long loop L3 [90].
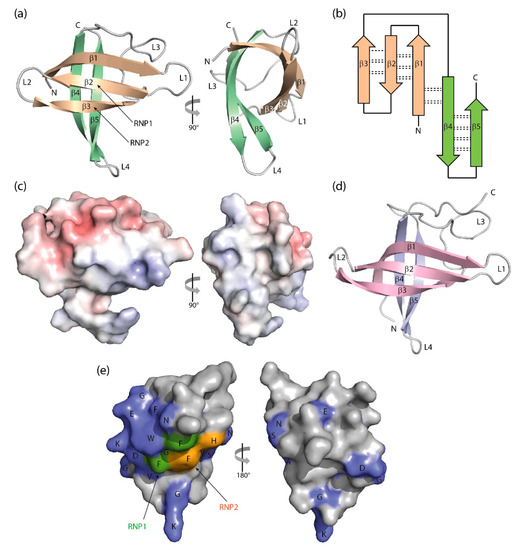
Figure 23.
Three-dimensional structure of cold-shock proteins and domains determined at near-atomic resolution. (
a
) Cartoon drawing, (
b
) topology diagram with β-sheet stabilizing hydrogen bonds, and (
c
) electrostatic surface potential colored from red (−10 kT/e) to blue (+10 kT/e) of
Bc
CspB (PDB entry 1c9o). (
d
) Schematic drawing of the
X. tropicalis
LIN28 CSD (PDB entry 3ulj). Orthogonal views are presented in (
a
,
c
). (
e
) Conserved residues on the surface of
StCspE (PDB entry 3i2z). RNP1/RNP2, RNA-binding motifs [12]. Note the close structural similarity between the bacterial
CspE (PDB entry 3i2z). RNP1/RNP2, RNA-binding motifs [20]. Note the close structural similarity between the bacterial
BcCspB [22] and the eukaryotic LIN28B CSD [26], the separation of negative (red) and positive (blue) surface charge in (
CspB [97] and the eukaryotic LIN28B CSD [100], the separation of negative (red) and positive (blue) surface charge in (
c) and the asymmetric distribution of conserved residues over the CSP surface. Cartoon drawings were prepared with PyMOL [27], the topology diagram is based on PDBsum [28], and the electrostatic surface was calculated with the Adaptive Poisson-Boltzmann Solver (APBS) plugin [29] of PyMOL.
) and the asymmetric distribution of conserved residues over the CSP surface. Cartoon drawings were prepared with PyMOL [101], the topology diagram is based on PDBsum [102], and the electrostatic surface was calculated with the Adaptive Poisson-Boltzmann Solver (APBS) plugin [103] of PyMOL.