Periodontitis, an inflammatory condition that affects the structures surrounding the tooth eventually leading to tooth loss, is one of the two biggest threats to oral health.
- Epstein–Barr virus
- periodontitis
- detection methods
- EBER-ISH
- PCR-based methods
- immunohistochemistry
- immunophenotyping
1. Introduction
Clinically periodontitis is defined as a chronic multifactorial inflammatory disease characterized by the progressive destruction of the tooth-supporting apparatus. The disease of periodontitis is portrayed by three factors: 1. the loss of periodontal-tissue support manifested through clinical attachment loss and radiographically assessed alveolar bone loss; 2. the presence of periodontal pockets (PP) and 3. gingival bleeding [1]. The periodontal disease initiates as gingivitis (inflammation of the gingiva), which is highly widespread and readily reversible by effective oral hygiene. When left untreated, it may gradually progress to early-to-moderate periodontitis and irreversible advanced periodontitis [2]. Periodontitis, along with dental caries, is considered one of the two biggest global oral health burdens [3]. Beyond oral health, growing evidence in the literature supports the direct and indirect impact of periodontitis on the overall health and development of extraoral pathologies. Periodontitis has been associated with seemingly unrelated systemic diseases such as diabetes, cardiovascular diseases and stroke, adverse pregnancy outcomes, respiratory diseases, dementia, Alzheimer’s disease, rheumatoid arthritis and different types of cancers [4,5][4][5]. However, it remains to be further scrutinized how specific periodontal pathogens contribute to the development of systemic diseases. However, as a minimum, the detection of periodontal pathogens in the oral cavity may be used as an attractive tool for the diagnosis of non-oral inflammatory systemic diseases.
There is a long history of the search for etiological agents of periodontitis and different hypotheses of etiopathogenesis have been proposed. Periodontitis was thought to be (i) an infection caused by bacteria; (ii) a specific bacterial infection; (iii) a biofilm infection; (iv) a specific plaque; (v) result from dysbiosis; (vi) caused by complex interactions among bacteria–host–environmental factors and (vii) a viral-bacterial infection (reviewed in [5,6][5][6]). One specific recent hypothesis of interest is based on the herpesvirus–pathogenic bacteria–host response axis in which herpesviral–bacterial interactions assume a major etiopathogenic role [5,7,8,9][5][7][8][9]. This infectious disease model for periodontitis development proposes that bacteria initiate the gingival inflammation triggering further influx and propagation of herpesviruses. Next, a herpesvirus active infection in the periodontium hinders the local immune defenses, thereby permitting the overgrowth of periodontopathic bacteria. In a two-way interaction, the virulence factors of periodontopathic bacteria reactivate latent herpesviruses and augment the infection. Reactivated periodontopathic herpesviruses and bacteria also modulate host immune reactions and provoke tissue destruction as a result of immunopathologic responses leading to the progression of the disease. In particular, among herpesviruses, human cytomegalovirus (HCMV) and Epstein–Barr virus (EBV) have been closely associated with severe types of periodontitis [10]. The main focus of the discussion in this review is EBV.
EBV belongs to the family of human gamma herpesviruses (systematic name human herpesvirus 4—HHV-4) and is one of the most ubiquitous and successfully adapted human pathogens that are found in approximately 95% of the total human population. EBV can infect a wide variety of cells and tissues, mostly B cells, nasopharynx and oropharynx squamous epithelial cells (ECs), thyroid glandular ECs, salivary and stomach glands and, occasionally, T cells, smooth muscle cells and follicular dendritic cells [11]. EBV has been associated with an extended list of diseases, from transient benign infections to aggressive malignancies. EBV is best known as the causative agent of infectious mononucleosis and has been implicated in several oral pathologies, such as oral hairy leukoplakia (OHL), oral lichen planus, Sjogren’s syndrome and periodontitis [9]. It is a known carcinogen implicated in the etiology of several malignancies of both lymphoid and epithelial origin [12]. Infection of B cells with EBV has been linked to Burkitt’s lymphoma, Hodgkin lymphoma and post-transplant lymphoproliferative disorders; infection of ECs is implicated in nasopharyngeal cancer, gastric cancer and breast cancer. Furthermore, a recent study suggested the association of EBV with seven different autoimmune diseases—multiple sclerosis, rheumatoid arthritis, inflammatory bowel disease, type 1 diabetes, juvenile idiopathic arthritis and celiac disease [13]. To confirm the etiopathogenic role of EBV in a disease, a better understanding of the EBV biology and molecular mechanism of the associated disease is required.
EBV is an enveloped DNA virus that has approximately 172 kb double-stranded DNA (dsDNA) genome encoding genes for latent and lytic infection. This virus was the first of the herpesviruses to be completely sequenced [14] [14] to identify over 80 protein-coding open reading frames and around 30 different non-coding RNAs (ncRNAs) [15].
Latent EBV infection allows for long-term viral persistence in the host, owing to tight control of viral gene expression to reduce the antiviral immune recognition. During latent infection, only several different types of RNAs and proteins are expressed. They include ncRNAs (EBV-encoded RNA 1 (EBER1) and EBER2, microRNAs (miRNA), EBV-stable intronic-sequence RNAs (EBV-sisRNA), EBV small nucleolar RNAs (EBV-snoRNA) and RPMS1 messenger RNA), six nuclear proteins (EBV nuclear antigen 1 (EBNA1), EBNA2, EBNA3A, EBNA3B, EBNA3C and EBNA5) and three latent membrane proteins (LMP1 and LMP2A-B) [11,15][11][15].
Upon lytic reactivation, EBV genes are sequentially expressed in immediate-early (IE), early (E) and late (L) states. Switch from latent to lytic state is triggered by expression of two IE viral transcription factors, the master regulator ZEBRA (also known as BZLF1, Zta, EB1 or Z) and Rta (BRLF1 or R). ZEBRA and Rta individually or cooperatively activate a subset of E genes many of which encode proteins involved in viral lytic DNA replication, such as the single-stranded DNA-binding protein (BALF2) and five replication enzymes and coenzymes, namely the helicase (BBLF4), primase (BSLF1), primase-associated factor (BBLF2/3), DNA polymerase (BALF5) and DNA polymerase processivity factor (BMRF1) [16]. For more details of E gene products, the reader is referred to the discussion in Kenney, 2007 [17]. Viral DNA replication is followed by expression of EBV’s L genes, which code for viral structural proteins (major capsid protein p160 (BcLF1) and three small capsid proteins, p18, p23 and p40 (BFRF3, BLRF2 and BdRF1)), glycoproteins (gp350/220 (BLLF1), gH (gp85; BXLF2), gp42 (BZLF2), etc.) and tegument proteins and viral interleukin 10 (vIL-10; BCLF1) [16,17,18][16][17][18]. All these latent and lytic determinants (the whole list is out of the scope of this review) are potentially useful for EBV detection and objective diagnosis.
Accurate laboratory tests to detect EBV are needed for purposes of basic and epidemiologic research and clinical management for different diseases. Biochemical, serological, immunological, histological, cytological and molecular detection methods of EBV have been used in the diagnosis and monitoring of patients with EBV-associated diseases [19]. The development of advanced laboratory methods allows timely and accurate diagnosis of clinical manifestations, which, in turn, may contribute to the prognosis and successful treatment. The identification of a suitable methodology that links EBV with different diseases will also advance our understanding of the molecular mechanisms underlying the onset and progression of EBV-associated diseases. (Figure 1)
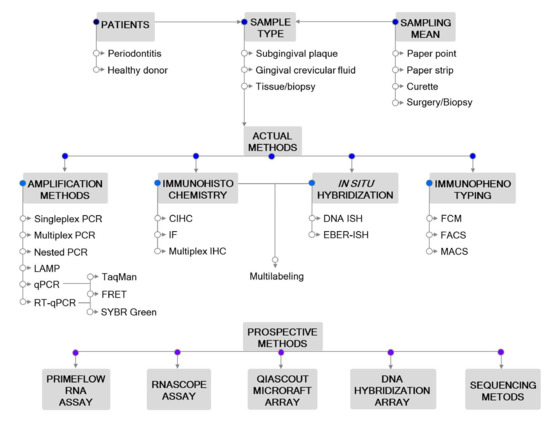
Figure 1. Outline of the methodological approaches for Epstein–Barr virus (EBV) detection in periodontitis. Actual and prospective methods are listed. Abbreviations: CIHC, chromogenic immunohistochemistry; EBER—EBV-encoded RNA; FACS—fluorescence-activated cell sorting; FCM—flow cytometry; FRET—fluorescence resonance energy transfer; IF—immunofluorescent detection; IHC—immunohistochemistry; ISH—in situ hybridization; LAMP—loop-mediated isothermal amplification; MACS—magnetic-activated cell sorting; PCR—polymerase chain reaction; qPCR—real-time quantitative PCR; RT-qPCR—reverse transcription qPCR.
2. Sampling
The physical presence of EBV in periodontal lesions suggests that EBV may be implicated in the etiopathogenesis of periodontitis. As such, samples need to be taken from the periodontal environment. The sample types and sample extraction methods may influence the identification and enumeration of microbes. The most eligible articles studying the association of EBV with periodontitis used subgingival plaque (SbgP), gingival crevicular fluid (GCF) and tissue/biopsy samples as sample type/sampling location [20], and curette, paper point, paper strip and surgery/biopsy as sample-extracting methods [21]. There is also a good deal of literature retrieving EBV from peripheral blood and saliva of periodontitis patients. Considering EBV is ubiquitous in the human population, the blood will not be reviewed here as a sample relevant to periodontitis, consequently, the serological tests are not discussed in this review. Though salivary EBV load may be very relevant to periodontitis, in this review, the saliva as a sample and salivary EBV load detection methods are not discussed either, because EBV is ubiquitous in humans, it is transmitted through saliva and EBV DNA is commonly detected also in the saliva from healthy adults [22].
The inspection of the subgingival plaque occupying the PP is considered the gold standard in studying periodontitis-associated microbial communities [23]. As might be expected, site-specific, intra- and interindividual variations of SbgP profiles may occur. In general, several paired SbgP samples are collected from shallow (healthy) and deep (diseased) sites from the same patient. The single-site analysis is preferential, but for practical and economic reasons, pooled SbgP samplings have often been performed [23]. Paper points are widely used for the collection of SbgP. Generally, the area of the collection is isolated with cotton rolls and air-dried to avoid contamination with saliva, then the supragingival plaque and calculus are carefully removed with a scaler to ensure the collection of only the subgingival material [24]. The color-coded paper points of specific sizes are inserted into the base of the PP, left in place for a certain duration and eluted. Basic parameters such as the origin of the paper points (manufacturer/supplier), the ISO size, probing (sampling) time and elution time may influence the optimum conditions for the microbiological sampling of PPs [25]. Samples can be collected as single (one paper point into the PP of each tooth), pooled (several paper points into the PPs of several teeth) or parallel (several paper points at one tooth) samples [26].
Curettes are also commonly used for sampling of subgingival specimens. After isolating the area with cotton rolls, a sterile curette tip is gently introduced through the pocket orifice into the bottom of the pocket and then removed with slight pressure against the tooth in a single vertical stroke to obtain the subgingival material [27,28][27][28].
In periodontitis, gingival crevicular fluid is an inflammatory exudate comprised of host-related substances, and from supra- and subgingival located microbes, thus, the analysis of GCF has become more and more important in the diagnosis of periodontitis [29]. Paper strips are used for GCF sample collection. The paper strip is inserted into the gingival crevice (intracrevicular method) or overlaid on the gingival crevice region (extracrevicular method). The intracrevicular method is subdivided to (i) superficial, when the strip is inserted just at the entrance of the crevice or PP and (ii) deep, when the strip is inserted to the base of the pocket or until minimum resistance is felt [30].
Tissue/biopsy specimens are obtained by periodontal surgery, which contain gingival tissue located adjacent to the PP. Careful dissection of the surgical piece can enrich the biopsy specimen with periodontal tissue attached to the tooth while removing the more distant gingival conjunctive areas. Dependent on the EBV detection method, the tissue specimens can further undergo cell dissociation, homogenization or fixation for isolation of macromolecules or histological analyses.
3. Polymerase Chain Reaction (PCR)-Based Detection Methods
PCR-based detection and quantification of EBV nucleic acids in body fluids and tissues have been used in the diagnosis and monitoring of EBV-associated diseases [19]. Extensive literature exists describing the application of PCR-based methods to identify and quantify EBV in periodontitis, which has targeted different genomic regions of EBV and applied different types of PCR methods. While most studies refer to the detection of the viral genomic DNA suitable to estimate the amounts of viruses in a specimen, a few other studies have focused on the detection of viral transcripts that can be more related to stages of viral replication in infected tissues. Specific characteristics and outcomes of recent studies (2010–2020) are summarized in Table 1. The large majority of these studies ascertain a strong association of EBV with periodontitis and its severity, indicating that EBV may serve as an etiopathogenic factor in periodontal diseases.
Table 1. Characteristics of the studies using nucleic acid amplification-based detection methods.
| Study | Periodontitis Type | Sample Size | Sample Type | Sampling Type | Amplification Type | Target | EBV Occurrence | Main Findings | |
|---|---|---|---|---|---|---|---|---|---|
| [38] | ApP | 40 ApP | PApT | Curette | Nested PCR (DNA) RT (RNA) + Nested PCR (cDNA) |
BamHI W (DNA) EBNA2 (RNA) |
29 ApP DNA |
20 ApP mRNA |
EBV infection is a frequent event in ApP. |
| 40 HC | PT | NA | 1 HC DNA |
1 HC mRNA |
|||||
| [37] | CP | 40 CP: 40 SS + 40 DS |
SbgP | Curette | Nested PCR (DNA) AGE |
EBNA2 | 4 SS + 29 DS | Significant association of EBV1 and CP. Association between EBV1 and periodontopathic bacteria. | |
| 40 HC | 1 HC | ||||||||
| [31] | AgP, CP | 20 AgP | SbgP | Curette | PCR (DNA) AGE |
EBNA2 | 9 AgP | Significantly higher prevalence of EBV1 in AgP and CP subjects compared to HCs. | |
| 20 CP | 5 CP | ||||||||
| 20 HC | 0 HC | ||||||||
| [33] | AgP, CP | 10 patients: 25 AgPS + 25 CPS 25 HS |
SbgP | Curette | Multiplex PCR (DNA) | ||||
]. EBNA1 was expressed at the highest and very similar levels to those measured in EBV-infected cell lines. Moreover, the EBNA1 expression level was correlated with the severity of the CP. On the other side, the IE viral transactivator BZLF1, known to induce the EBV lytic cycle, was also expressed in CP samples but at a level lower than that observed in the EBV-producing cell line. Overall, their conclusions derived from RT-qPCR analysis were that EBV-infected periodontal cells were likely in a state of latent EBV infection and that the level of EBV infection correlated with disease severity.
4. Immunohistochemistry (IHC)
To identify the precise cellular location of EBV, morphology-based techniques are used. IHC may be applied to confirm the presence, distribution, localization of EBV in the cells/tissues and distinguish latent from lytic infection based on protein expression profiles. IHC for EBV detection involves the staining of key EBV proteins such as EBNA1, EBNA2, LMP1, LMP2A and BZLF1 [63]. Commercial antibodies to EBV for IHC assays are available. There are also automated and standardized procedures routinely and widely used in pathology laboratories to detect EBV proteins in tissue specimens, such as FLEX monoclonal mouse anti-Epstein–Barr virus, LMP, Clones CS.1–4 (DAKO), which are used together with Autostainer Link instruments.
IHC procedures are performed on formalin-fixed, paraffin-embedded (FFPE) tissue sections of periodontal biopsies and cytological preparations from the periodontal environment. A standard IHC protocol is a multistep procedure involving deparaffinization/rehydration, heat- or proteolytic-induced antigen retrieval, blocking of non-specific staining, permeabilization, immunostaining with a primary antibody specific to a target antigen, incubation with labeled secondary antibody and detection. IHC allows for chromogenic (chromogenic immunohistochemistry—CIHC) and fluorescent (immunofluorescent—IF) detection types. For CIHC detection, the antibody is conjugated to an enzyme (such as horseradish peroxidase (HRP) or alkaline phosphatase (AP)), which converts a substrate (such as 3,3′-diaminobenzidine (DAB) or 3-amino-9-ethylcarbazole (AEC)) into a colored precipitate at the antigen site. For IF detection, the fluorophore (such as Alexa Fluor family dyes or fluorescein isothiocyanate (FITC)) conjugated antibody is excited by and emits light at specific wavelengths. Following the immunostaining, counterstaining with hematoxylin (chromogenic detection) or with DAPI (fluorescence detection) is performed to contextualize the antigen of interest. After the completion of all staining, the tissue is mounted and visualized by a bright-field (CIHC) or fluorescence/confocal (IF) microscope.
Multiplex chromogenic and fluorescence immunohistochemistry has recently emerged as a potent tool for the simultaneous detection of multiple biological markers on a single tissue section using a consecutive or simultaneous staining approach [64]. Multiplexed strategies allow compiling maximal information per tissue section of a limited sample and to understand coexpression and colocalization of multiple targets within tissue architecture.
Though IHC is a sensitive, versatile technique with many applications, careful control selection and proper optimization of the protocol is required. Besides, because the evaluation of the staining intensity of IHC is subjective, the ambiguity in the evaluation of the results and inter- or intraobserver variability may be problematic [65].
IHC, multiplex IHC and combined IHC techniques were employed for EBV analysis in periodontitis research (Table 2). In this context, using the CIHC approach for LMP1 protein immunostaining Saboia-Dantas et al. observed EBV in 31% of apical periodontitis lesions obtained after teeth extraction [66]. Vincent-Bugnas et al. applied IF costaining of viral latent proteins LMP1 and LMP2, and junctional EC marker cytokeratin 19 (CK19) to detect latent EBV-infected periodontal ECs (pECs) in non-surgical liquid-based cytological samples derived from PPs of CP patients (Figure 2 [27]). They estimated that around 32% of the CK19+ cells were infected with EBV (LMP2+).
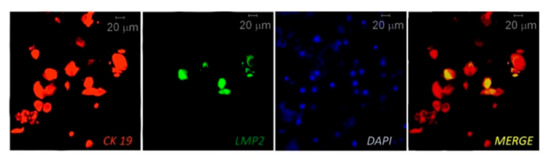
Figure 2. Immunofluorescent (IF) costaining of CK19 (junctional epithelial cell marker cytokeratin 19) and LMP2 (EBV latent membrane protein 2) to detect EBV-infected epithelial cells in samples taken from a periodontitis patient. The cell nuclei are counterstained with DAPI. Reprinted from [27].
Table 2. Characteristics of the studies using tissue-based detection methods.
| Study | Periodontitis Type | Sample Size | Sample Type | Sampling Type | Tissue-Based Detection Type |
Target | EBV Occurrence | Main Findings | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| [66] | ApP | 35 ApP | Apical lesion | Teeth extraction |
CIHC | LMP1 | 11 ApP | EBV occurrence in about 31% of ApP samples. | ||||||||
| [67] | ApP | 20 ApP | Apical lesion | Submarginal incision | EBER-ISH | EBER | 0 ApP | No signs of cells harboring EBV in 20 apical samples analyzed by EBER-ISH. | ||||||||
| [39] | CP | 41 SS + 56 DS | Gingival tissue | Flap surgery |
EBER-ISH + CIHC |
EBER CD19 |
EBER+ CD19+ |
Numerous CD19+ B cells infiltrated in the connective tissue subjacent to the gingival epithelium; numerous cells in the same location were EBER+. | ||||||||
| [27] | CP | 3 CP: 3 PP 3 CP: 3 PalS |
SbgP PalECs |
Curette, cytospin cuvette |
||||||||||||
| AGE | ||||||||||||||||
| LMP2 | 8 AgPS + 8 CPS | 2 HS | Significant association of EBV with CP and AgP. | |||||||||||||
| [39] | CP | 85 CP: 85 SS + 85 DS |
SbgP | Paper point |
Nested PCR (DNA) AGE |
EBNA2 | 41 SS + 56 DS | More frequent detection of EBV DNA in patients with DS than in those with SS or HCs. EBV DNA may serve as a pathogenic factor leading to CP. | ||||||||
| 20 HC: 40 HS |
18 HS | |||||||||||||||
| [56] | AgP | 65 AgP | SbgP | Paper point |
FRET qPCR (DNA) |
BRLF1 | 7 AgP | No association between EBV and AgP. | ||||||||
| 65 HC | 9 HC | |||||||||||||||
| [34] | AgP | 15 AgP | SbgP, IDPT | Curette | Multiplex PCR (DNA) AGE |
LMP2 | 10 AgP SbgP 11 AgP IDPT |
Significant prevalence of EBV in AgP compared to HCs. | ||||||||
| 15 HC | 1 HC SbgP 0 HC IDPT |
|||||||||||||||
| [27] | CP | 6 CP: 6 SS + 6 DS |
SbgP | Curette | SYBR Green RT-qPCR (RNA) |
EBNA1, EBNA2, LMP1, LMP2, BZLF1 |
6 SS + 6 DS: EBNA1 > EBNA2 ≥ LMP1 ≥ LMP2 ≥ BZLF1 |
EBV-specific latent (LMP1, LMP2, EBNA1 and EBNA2) and lytic (BZLF1) transcripts detected in all PP but not PalEC samples of CP patients. | ||||||||
| 3 CP: 3 PalS |
PalECs | 3 PalS: EBNA1 = EBNA2 = LMP1 = LMP2 = BZLF1 = 0 |
||||||||||||||
| 10 HC: 10 HS |
GS | EBNA1 | DS > SS > HS | EBNA1 transcripts detected 36- and 5-fold higher in DS and SS, respectively, compared to HS. | ||||||||||||
| [40] | ApP | 100 ApP | PApT | Curette | Nested PCR (DNA) PAGE |
EBNA2 | 76 ApP | Significant occurrence of EBV1 genotype in periapical lesions than in healthy pulps. | ||||||||
| 25 HC | PT | Endodontic file |
6 HC | |||||||||||||
| IF costaining | LMP1, | LMP2 | CK19 | 3 PP | 0 PalS |
Around 32% of the CK19+ epithelial cells infected with EBV (LMP2+). | ||||||||||
| EBER-ISH + CIHC |
EBER CK19 |
EBER+, CK19+ PP EBER-, CK19+ PalS |
EBER+ periodontal epithelial cells (pECs) were detected only in PP samples. | |||||||||||||
| 20 CP: 20 SS + 20 DS 10 HC: 10 HS |
SbgP GS |
Curette, cytospin cuvette |
EBER-ISH + CIHC |
EBER CK19 |
DS > SS > HS | Frequency of EBV+ pECs higher in deep pockets than in shallow pockets and healthy sites. A positive correlation between EBV infection and disease severity. | ||||||||||
| [68] | PApP | 9 PApP | 6 PApP | PApG | Endodontic surgery | EBER-ISH | CIHC | EBER | LMP1 | 6 PApP | 6PApP | EBER detected in the cytoplasm and nuclei of B cells and plasma cells (PC) in 66.7% of PApGs, but not in healthy gingival tissues. All EBER+ PApGs positive for LMP1. LMP-1-expressing cells localized in the same areas as EBER-expressing cells. |
||||
| 5 HC | 5 HC | Gingival tissue | Teeth extraction |
0 HC | 0 HC | |||||||||||
| [62] | CP (SvP) | 5 SvP | Gingival tissue | Surgery | EBER-ISH + | EBER | EBER+ | Numerous EBV-infected cells, mostly overlapping with CD138+ PCs. EBV-infected PCs formed high-density clusters along the periodontal epithelium associated with CD3+ T cells and CD20+ B cells. | ||||||||
| Multiplex CIHC | CD3, CD20, CD138, Kappa |
CD3+, CD20+, CD138+, Kappa | ||||||||||||||
| [32] | CP | 100 CP | SbgP | Curette | PCR (DNA) AGE |
LMP2 | 21 CP | Significantly higher levels of EBV in CP as compared to the healthy periodontium. | ||||||||
| 100 HC | 6 HC | |||||||||||||||
| + | ||||||||||||||||
| [28] | AgP | 15 AgP | SbgP | Curette | Hotstart PCR (DNA) AGE |
NA | 6 AgP | EBV occurrence comparable among AgP and HC groups. | ||||||||
| 15 HC | 1 HC | |||||||||||||||
| [57] | CP | 60 CP | Tissue | Surgery | TaqMan qPCR (DNA) |
NA | DS > SS | Observed EBV in tissue samples from deep and shallow PPs. Quantification of EBV is high in periodontal tissue samples of severe CP. | ||||||||
| [58] | CP | 25 CP: 25 SS + 25 DS |
SbgP | Paper point |
SYBR Green qPCR (DNA) |
BNRF1 | 10 SS + 20 DS | Significantly high EBV DNA in DS than in SS of CP patients and HS of HCs. Association between EBV DNA, P. gingivalis and CP. | ||||||||
| 13 HC: 26 HS |
13 HS | |||||||||||||||
| [35] | CP | 40 CP | GCF | Paper strip |
Multiplex PCR (DNA) AGE |
LMP2 | 25 CP | Significantly higher prevalence of EBV in GCF of CP patients than in HCs. Strong association between EBV and CP. | ||||||||
| 20 HC | 2 HC | |||||||||||||||
| [36] | CP (MiP, MdP, SvP) | 100 MiP + 100 MdP + 100 SvP | SbgP | Curette, paper point |
Multiplex PCR (DNA) AGE |
LMP2 | 25 MiP + 20 MdP + 47 SvP |
Significant association between EBV and CP, and the severity of the disease. | ||||||||
| 300 HC | 0 HC | |||||||||||||||
| [54] | AgP | 17 AgP | SbgP | Paper point |
LAMP (DNA) AGE + TA |
BamHI W | 64.7% AgP | No significant association between EBV1 and AgP. Highest risk of AgP when A. actinomycetemcomitans and EBV1/HCMV are together. | ||||||||
| 17 HC | 47.1% HC | |||||||||||||||
| [59] | GAP | 165 GAP: 165 AS + 165 n-AS |
SbgP | Paper point |
qPCR (DNA) AGE |
EBNA1 | 23 AS + NA n-AS | EBV association with A. actinomycetemcomitans. Although the presence of EBV (herpesvirus in general) is not necessary for the progression of GAP, it can facilitate it, possibly by promoting pathogenicity and virulence of periodontopathic bacteria in a virus and bacterial species-dependent manner. | ||||||||
| [60] | AgP, CP | 18 AgP +12 CP | SbgP | Curette | TaqMan qPCR (DNA) |
NA | 19 (AgP + CP) | Significant presence of EBV in periodontitis sites as compared to healthy sites. Positive correlation of EBV with P. gingivalis and T. forsythia. | ||||||||
| 30 HC | 3 HC | |||||||||||||||
| [61] | AgP, ApP | 22 AgP + 3 ApP | SbgP | Paper point |
TaqMan qPCR (DNA) |
EBNA1 | 16 AgP + 3 ApP | Prevalence and copy number of EBV significantly higher in periodontitis patients than in healthy controls. | ||||||||
| 25 HC | 4 HC | |||||||||||||||
| [62] | CP (MdP, SvP) | 20 patients: 9 MdP + 11 SvP |
SbgP | Curette | TaqMan EBV R-GENE qPCR (DNA) |
BXLF1 | 0–9861.14 × 102 copies/µg |
Different levels of EBV occurrence in CP patients. | ||||||||
| [41] | AgP, CP | 57 AgP | Tissue | Surgery | Nested PCR (DNA) AGE SYBER Green qPCR (DNA) |
EBNA2 BALF5 |
25 AgP | 4.41–7.01 log10 copies/g AgP | Significant occurrence of EBV in the AgP and CP groups compared to the HC. Significant association between EBV load and periodontitis. |
|||||||
| 59 Cp | 28 CP | 5.06–7.31 log10 copies/g CP | ||||||||||||||
| 43 HC | 5 HC | 4.57–5.21 log10 copies/g HC | ||||||||||||||
Abbreviations: Periodontitis type: AgP—aggressive periodontitis; ApP—apical periodontitis; CP—chronic periodontitis; HC—healthy control; GAP—generalized aggressive periodontitis; MdP—moderate periodontitis; MiP—mild periodontitis; SvP—severe periodontitis. Sample type: GCF—gingival crevicular(/periodontal pocket) fluid; GS—gingival sulcus; IDPT—interdental papilla tissue; PApT—periapical tissue; PalECs—palatal epithelial cells; PP—periodontal pocket; PT—pulp tissue; SbgP—subgingival plaque. Site type: AgPS—aggressive periodontitis site; AS—active site; CPS—chronic periodontitis site; DS—deep site; HS—healthy site; n-AS—non-active site; PalS—palatal site; SS—shallow site. Amplification type: FRET—fluorescence resonance energy transfer; PCR—polymerase chain reaction: qPCR—real-time quantitative PCR; RT—reverse transcription reaction; RT-qPCR—reverse transcription qPCR. Amplicon detection type: AGE—agarose gel electrophoresis; PAGE—polyacrylamide gel electrophoresis; TA—turbidity assay. NA—not available.
Conventional singleplex PCR was applied to amplify and detect a single target gene of EBV, such as EBNA2 [31] and LMP2 [32], while multiplex PCR was used to simultaneously amplify target sequences of several periodontopathic bacteria and herpesviruses [33,34,35,36][33][34][35][36]. Another PCR technique, the nested PCR, was implemented to increase the specificity of EBV DNA amplification and reduce the non-specific amplification by the involvement of two sets of primers (outer and inner pairs) for the same target [37,38,39,40,41][37][38][39][40][41]. Additionally, the nested PCR is more efficient in detecting low viral loads [42]. Typically, the amplicons generated via these PCR methods are subsequently size-fractioned and detected by agarose (AGE) or polyacrylamide gel electrophoresis (PAGE) (Table 1), or else, restriction fragment length polymorphism analysis (RFLP) with endonucleases followed by AGE can be applied. For example, Afa I digests the 497 bp amplicon of EBV1 in 355 bp and 142 bp fragments, while Stu I digests the 165 bp amplicon of EBV2 in 118 bp and 47 bp fragments, which can be visualized with AGE [43].
Due to EBV ubiquity and life-long persistent infection, simply detecting it is insufficient to diagnose EBV association with the disease [44]. Quantitative measurement of the EBV genome is necessary to distinguish between low-level EBV infection in healthy carriers and high levels typical to EBV-associated diseases [45]. Real-time quantitative PCR (further named qPCR) is the main method for modern EBV viral load measurement, which also eliminates post-PCR manipulations. Real-time qPCR is based on the amplification of a conserved sequence (typically around 100 bp) using either a fluorescent probe (e.g., TaqMan probe and fluorescence resonance energy transfer (FRET) hybridization probe) or an intercalating dye (e.g., SYBR Green) coupled with real-time laser scanning to quantify the target DNA against serial dilutions of known EBV DNA content [11,45][11][45]. Quantified EBV genomic DNA (gDNA) sources are now commercially available that may be used for assay calibration [45].
In the TaqMan system, in addition to the two amplification primers used in conventional PCR, a dual-labeled fluorogenic hybridization probe is used. The probe hybridizes specifically in the DNA target region between the two PCR primers. One fluorescent dye serves as a reporter and its emission is quenched by the second fluorescent dye. Nuclease degradation of the hybridization probe by Taq DNA polymerase releases quenching of the reporter fluorescence, resulting in an increase in peak fluorescence [44,46][44][46]. The principle of FRET hybridization is based on the hybridization of two single-stranded, sequence-specific, fluorescent-labeled oligonucleotides (with donor and acceptor dyes) to the target sequence in close proximity in a head-to-tail orientation. The energy absorbed by the donor fluorophore is transferred to the acceptor fluorophore, which then emits fluorescence (FRET) [47]. In the SYBR Green system, a green dye is used as a marker for product accumulation, which intercalates into dsDNA as PCR products accumulate [45,48][45][48]. The SYBR Green system is less expensive but less specific in comparison with probe strategies.
Real-time qPCR is considered a sensitive, reliable, stringent, simple, specific, precise and fast method [11]. Since nucleic acid amplification and detection occurs in the same sealed tube, the risk of amplicon contamination is negligent compared with conventional PCR methods. Due to advanced instrumentation, the real-time qPCR testing is much simpler to perform and the test results are acquired much faster [49]. Currently, several real-time qPCR-based EBV detection and quantification kits are commercially available, such as EBV R-GENE (bioMérieux), EBV ELITe MGB (ELITechGroup), artus EBV PCR Kits (QIAGEN), etc. However, attention should be paid when comparing the data of different studies [50]. The source of deviation could be the units of measurement (copies per milliliter, copies per microgram of DNA, copies per positive cell) or the EBV targets (LMP2, BKRF1 or BamHI W (EBNA1), BNRF1 (membrane protein), BXLF1 (thymidine kinase), BZLF1, BALF5 or BHRF1 (transmembrane protein), etc.).
EBV DNA can be detected with high specificity, sensitivity and rapidity on par with the real-time qPCR method utilizing the loop-mediated isothermal amplification (LAMP) method. The LAMP reaction requires a DNA polymerase with strand displacement activity and a set of four specially designed inner and outer primers that recognize a total of six distinct sequences within the target DNA. Iwata et al. (2006) designed primers for the EBV LAMP assay based on BamHI W gene sequences [51]; Liu et al. (2013) later designed an extended set of LAMP primers for latent (EBNA1, EBNA2, LMP1 and LMP2A) and lytic (BZLF1) transcripts [52]. During LAMP reaction specific DNA targets are amplified at 63–65 °C, without thermocycling, accumulating 109 copies of the target in less than an hour. The final products of LAMP are stem-loop DNAs with several inverted repeats of the target DNA and cauliflower-like structures with multiple loops. LAMP amplicon product is further detected by turbidity assay (TA) of the white precipitate of magnesium pyrophosphate and/or AGE. The reaction is described in detail in [51,53][51][53]. Elamin et al. used this technique to assess the presence of putative periodontopathic bacteria (Aggregatibacter actinomycetemcomitans, Porphyromonas gingivalis, Tannerella forsythia and Treponema denticola) and two periodontal herpesviruses (EBV1 and HCMV) in individuals with aggressive periodontitis [54]. Though they reported no significant association between EBV1 and the disease, the highest risk of aggressive periodontitis was observed when A. actinomycetemcomitans was detected together with EBV1 and/or HCMV.
Using a sensitive and reproducible reverse transcription qPCR (RT-qPCR) method different transcripts of EBV can be detected and quantified to distinguish distinct states of latent or lytic EBV infection or closely monitor reactivation of EBV. In theory, RT-qPCR differs from qPCR only by the addition of a preliminary step, the initial complementary DNA (cDNA) synthesis from an RNA template by an RNA-dependent DNA polymerase (reverse transcriptase). After the RT reaction, suitable detection chemistry to report the presence of PCR amplicons, an instrument to monitor the amplification in real-time and appropriate software for quantitative analysis are required [55]. From the list of recent studies (Table 1), Hernádi et al. used the RT reaction to convert the EBNA2 messenger RNA into cDNA followed by nested PCR to detect that EBNA2 expression was significantly more frequent in apical periodontitis lesions as compared to healthy controls [38]. They concluded that EBV infection was a frequent event in apical periodontitis and that symptoms were likely to occur if the lesion is aggravated with active EBV infection. Vincent-Bugnas and coauthors used the sensitive RT-qPCR technique and observed that EBV latent (EBNA1, EBNA2, LMP1 and LMP2) transcripts were detectable in all PP samples of chronic periodontitis (CP) patients, which were within the range expressed by EBV-infected cell lines [27
Abbreviations: Periodontitis type: ApP—apical periodontitis; CP—chronic periodontitis; PApP—periapical periodontitis; HC—healthy control; SvP—severe periodontitis. Sample type: GS—gingival sulcus; PApG—periapical granuloma; PalECs—palatal epithelial cells; SbgP—subgingival plaque. Site type: DS—deep site; HS—healthy site; PalS—palatal site; PP—periodontal pocket; SS—shallow site. Tissue-based analysis type: CIHC—chromogenic immunohistochemistry; EBER-ISH—EBV-encoded RNA in situ hybridization; IF—immunofluorescent staining.
References
- Papapanou, P.N.; Sanz, M.; Buduneli, N.; Dietrich, T.; Feres, M.; Fine, D.H.; Flemmig, T.F.; Garcia, R.; Giannobile, W.V.; Graziani, F.; et al. Periodontitis: Consensus Report of Workgroup 2 of the 2017 World Workshop on the Classification of Periodontal and Peri-Implant Diseases and Conditions: Classification and Case Definitions for Periodontitis. J. Clin. Periodontol. 2018, 45, S162–S170. [Google Scholar] [CrossRef] [PubMed]
- Kinane, D.F.; Stathopoulou, P.G.; Papapanou, P.N. Periodontal Diseases. Nat. Rev. Dis. Primer 2017, 3, 17038. [Google Scholar] [CrossRef] [PubMed]
- WHO. What Is the Burden of Oral Disease? Available online: https://www.who.int/oral_health/disease_burden/global/en/ (accessed on 21 October 2020).
- Bui, F.Q.; Almeida-da-Silva, C.L.C.; Huynh, B.; Trinh, A.; Liu, J.; Woodward, J.; Asadi, H.; Ojcius, D.M. Association between Periodontal Pathogens and Systemic Disease. Biomed. J. 2019, 42, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Feng, P.; Slots, J. Herpesvirus-Bacteria Synergistic Interaction in Periodontitis. Periodontology 2000 2020, 82, 42–64. [Google Scholar] [CrossRef] [PubMed]
- Teles, R.; Teles, F.; Frias-Lopez, J.; Paster, B.; Haffajee, A. Lessons Learned and Unlearned in Periodontal Microbiology. Periodontology 2000 2013, 62, 95–162. [Google Scholar] [CrossRef]
- Slots, J. Herpesviral-Bacterial Synergy in the Pathogenesis of Human Periodontitis. Curr. Opin. Infect. Dis. 2007, 20, 278–283. [Google Scholar] [CrossRef]
- Slots, J. Periodontal Herpesviruses: Prevalence, Pathogenicity, Systemic Risk. Periodontology 2000 2015, 69, 28–45. [Google Scholar] [CrossRef]
- Tonoyan, L.; Vincent-Bugnas, S.; Olivieri, C.-V.; Doglio, A. New Viral Facets in Oral Diseases: The EBV Paradox. Int. J. Mol. Sci. 2019, 20, 5861. [Google Scholar] [CrossRef]
- Kamma, J.J.; Slots, J. Herpesviral–Bacterial Interactions in Aggressive Periodontitis. J. Clin. Periodontol. 2003, 30, 420–426. [Google Scholar] [CrossRef]
- Abusalah, M.A.H.; Gan, S.H.; Al-Hatamleh, M.A.I.; Irekeola, A.A.; Shueb, R.H.; Yean, C.Y. Recent Advances in Diagnostic Approaches for Epstein–Barr Virus. Pathogens 2020, 9, 226. [Google Scholar] [CrossRef]
- Khan, G.; Hashim, M.J. Global Burden of Deaths from Epstein-Barr Virus Attributable Malignancies 1990–2010. Infect. Agent. Cancer 2014, 9, 38. [Google Scholar] [CrossRef] [PubMed]
- Harley, J.B.; Chen, X.; Pujato, M.; Miller, D.; Maddox, A.; Forney, C.; Magnusen, A.F.; Lynch, A.; Chetal, K.; Yukawa, M.; et al. Transcription Factors Operate across Disease Loci, with EBNA2 Implicated in Autoimmunity. Nat. Genet. 2018, 50, 699–707. [Google Scholar] [CrossRef] [PubMed]
- Robertson, E.S. (Ed.) Epstein-Barr Virus; Caister Academic Press: Wymondham, UK, 2005; ISBN 978-1-904455-03-5. [Google Scholar]
- Skalsky, R.L.; Cullen, B.R. EBV Noncoding RNAs. Curr. Top. Microbiol. Immunol. 2015, 391, 181–217. [Google Scholar] [CrossRef] [PubMed]
- McKenzie, J.; El-Guindy, A. Epstein-Barr Virus Lytic Cycle Reactivation. In Epstein Barr Virus Volume 2: One Herpes Virus: Many Diseases; Münz, C., Ed.; Current Topics in Microbiology and Immunology; Springer International Publishing: Cham, Switzerland, 2015; pp. 237–261. ISBN 978-3-319-22834-1. [Google Scholar]
- Kenney, S.C. Reactivation and Lytic Replication of EBV. In Human Herpesviruses: Biology, Therapy, and Immunoprophylaxis; Arvin, A., Campadelli-Fiume, G., Mocarski, E., Moore, P.S., Roizman, B., Whitley, R., Yamanishi, K., Eds.; Cambridge University Press: Cambridge, UK, 2007; ISBN 978-0-521-82714-0. [Google Scholar]
- Young, L.S.; Arrand, J.R.; Murray, P.G. EBV Gene Expression and Regulation. In Human Herpesviruses: Biology, Therapy, and Immunoprophylaxis; Arvin, A., Campadelli-Fiume, G., Mocarski, E., Moore, P.S., Roizman, B., Whitley, R., Yamanishi, K., Eds.; Cambridge University Press: Cambridge, UK, 2007; ISBN 978-0-521-82714-0. [Google Scholar]
- Ayee, R.; Ofori, M.E.O.; Wright, E.; Quaye, O. Epstein Barr Virus Associated Lymphomas and Epithelia Cancers in Humans. J. Cancer 2020, 11, 1737–1750. [Google Scholar] [CrossRef]
- Zhu, C.; Li, F.; Wong, M.C.M.; Feng, X.-P.; Lu, H.-X.; Xu, W. Association between Herpesviruses and Chronic Periodontitis: A Meta-Analysis Based on Case-Control Studies. PLoS ONE 2015, 10, e0144319. [Google Scholar] [CrossRef] [PubMed]
- Gao, Z.; Lv, J.; Wang, M. Epstein–Barr Virus Is Associated with Periodontal Diseases. Medicine 2017, 96, e5980. [Google Scholar] [CrossRef]
- Chen, T.; Hudnall, S.D. Anatomical Mapping of Human Herpesvirus Reservoirs of Infection. Mod. Pathol. 2006, 19, 726–737. [Google Scholar] [CrossRef]
- Belstrøm, D.; Sembler-Møller, M.L.; Grande, M.A.; Kirkby, N.; Cotton, S.L.; Paster, B.J.; Holmstrup, P. Microbial Profile Comparisons of Saliva, Pooled and Site-Specific Subgingival Samples in Periodontitis Patients. PLoS ONE 2017, 12, e0182992. [Google Scholar] [CrossRef]
- Gokhale, S.; Padhye, A.; Sumanth, S. Bactericidal Effect of Nd: YAG Laser in an In Vitro Tissue Model—A Light Microscopic Evaluation. J. Oral Laser Appl. 2010, 10, 17–22. [Google Scholar]
- Hartroth, B.; Seyfahrt, I.; Conrads, G. Sampling of Periodontal Pathogens by Paper Points: Evaluation of Basic Parameters. Oral Microbiol. Immunol. 1999, 14, 326–330. [Google Scholar] [CrossRef]
- Santigli, E.; Koller, M.; Klug, B. Oral Biofilm Sampling for Microbiome Analysis in Healthy Children. JoVE J. Vis. Exp. 2017, e56320. [Google Scholar] [CrossRef] [PubMed]
- Vincent-Bugnas, S.; Vitale, S.; Mouline, C.C.; Khaali, W.; Charbit, Y.; Mahler, P.; Prêcheur, I.; Hofman, P.; Maryanski, J.L.; Doglio, A. EBV Infection Is Common in Gingival Epithelial Cells of the Periodontium and Worsens during Chronic Periodontitis. PLoS ONE 2013, 8, e80336. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Tapashetti, R.P.; Patil, S.R.; Kalra, S.M.; Bhat, G.K.; Guvva, S. Revelation of Viral–Bacterial Interrelationship in Aggressive Periodontitis via Polymerase Chain Reaction: A Microbiological Study. J. Int. Oral Health JIOH 2015, 7, 101–107. [Google Scholar]
- Guentsch, A.; Kramesberger, M.; Sroka, A.; Pfister, W.; Potempa, J.; Eick, S. Comparison of Gingival Crevicular Fluid Sampling Methods in Patients with Severe Chronic Periodontitis. J. Periodontol. 2011, 82, 1051–1060. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, G.S. Formation, Collection and Significance of Gingival Crevice Fluid. Periodontology 2000 2003, 31, 32–42. [Google Scholar] [CrossRef] [PubMed]
- Sharma, R.; Padmalatha, O.; Kaarthikeyan, G.; Jayakumar, N.D.; Varghese, S.; Sherif, K. Comparative Analysis of Presence of Cytomegalovirus (CMV) and Epsteinbarr Virus -1 (EBV-1) in Cases of Chronic Periodontitis and Aggressive Periodontitis with Controls. Indian J. Dent. Res. Off. Publ. Indian Soc. Dent. Res. 2012, 23, 454–458. [Google Scholar] [CrossRef] [PubMed]
- Joshi, V.M.; Bhat, K.G.; Katti, S.S.; Kugaji, M.S.; Ingalgi, P.S. Prevalence of Herpesvirus and Correlation with Clinical Parameters in Indian Subjects with Chronic Periodontitis. J. Contemp. Dent. Pract. 2015, 16, 915–920. [Google Scholar] [CrossRef]
- Das, S.; Krithiga, G.S.P.; Gopalakrishnan, S. Detection of Human Herpes Viruses in Patients with Chronic and Aggressive Periodontitis and Relationship between Viruses and Clinical Parameters. J. Oral Maxillofac. Pathol. 2012, 16, 203. [Google Scholar] [CrossRef]
- Dani, S.; Dwarakanath, C.; Alampalli, R.; Bhat, K.; An, S.; Gundannavar, G. Role of Herpes Simplex-1, Epstein Barr and Human Cytomegalo Viruses in Aggressive Periodontitis. Int. J. Dent. Res. 2013, 1, 19–24. [Google Scholar] [CrossRef]
- Shah, R.; Mehta, D.S. Prevalence of Herpesviruses in Gingivitis and Chronic Periodontitis: Relationship to Clinical Parameters and Effect of Treatment. J. Indian Soc. Periodontol. 2016, 20, 279–285. [Google Scholar] [CrossRef]
- Kazi, M.M.A.G.; Bharadwaj, R. Role of Herpesviruses in Chronic Periodontitis and Their Association with Clinical Parameters and in Increasing Severity of the Disease. Eur. J. Dent. 2017, 11, 299–304. [Google Scholar] [CrossRef] [PubMed]
- Chalabi, M.; Rezaie, F.; Moghim, S.; Mogharehabed, A.; Rezaei, M.; Mehraban, B. Periodontopathic Bacteria and Herpesviruses in Chronic Periodontitis. Mol. Oral Microbiol. 2010, 25, 236–240. [Google Scholar] [CrossRef] [PubMed]
- Hernádi, K.; Szalmás, A.; Mogyorósi, R.; Czompa, L.; Veress, G.; Csoma, E.; Márton, I.; Kónya, J. Prevalence and Activity of Epstein-Barr Virus and Human Cytomegalovirus in Symptomatic and Asymptomatic Apical Periodontitis Lesions. J. Endod. 2010, 36, 1485–1489. [Google Scholar] [CrossRef] [PubMed]
- Kato, A.; Imai, K.; Ochiai, K.; Ogata, Y. Higher Prevalence of Epstein–Barr Virus DNA in Deeper Periodontal Pockets of Chronic Periodontitis in Japanese Patients. PLoS ONE 2013, 8, e71990. [Google Scholar] [CrossRef]
- Jakovljevic, A.; Andric, M.; Knezevic, A.; Soldatovic, I.; Nikolic, N.; Karalic, D.; Milasin, J. Human Cytomegalovirus and Epstein-Barr Virus Genotypes in Apical Periodontitis Lesions. J. Endod. 2015, 41, 1847–1851. [Google Scholar] [CrossRef]
- Yu, T.; Pan, S.; Zhang, Y.; Pei, J.; Liu, J.; Xie, Y.; Feng, X. Occurrence and Quantification of Anelloviruses and Herpesviruses in Gingival Tissue in Chinese Shanghai Sub-Population. BMC Oral Health 2020, 20, 196. [Google Scholar] [CrossRef]
- Varghese, S. Role of Herpes Virus in Periodontal Diseases. IOSR J. Dent. Med. Sci. 2014, 13, 71–73. [Google Scholar] [CrossRef]
- Wu, Y.; Yan, J.; Chen, L.; Sun, W.; Gu, Z. Infection Frequency of Epstein-Barr Virus in Subgingival Samples from Patients with Different Periodontal Status and Its Correlation with Clinical Parameters. J. Zhejiang Univ. Sci. B 2006, 7, 876–883. [Google Scholar] [CrossRef]
- Kimura, H.; Ito, Y.; Suzuki, R.; Nishiyama, Y. Measuring Epstein–Barr Virus (EBV) Load: The Significance and Application for Each EBV-Associated Disease. Rev. Med. Virol. 2008, 18, 305–319. [Google Scholar] [CrossRef]
- Gulley, M.L.; Tang, W. Laboratory Assays for Epstein-Barr Virus-Related Disease. J. Mol. Diagn. 2008, 10, 279–292. [Google Scholar] [CrossRef]
- Lo, D.; Chan, L.; Lo, K.W.; Leung, S.; Zhang, J.; Chan, A.; Lee, J.; Hjelm, N.; Johnson, P.; Huang, D.P. Quantitative Analysis of Cell-Free Epstein-Barr Virus DNA in Plasma of Patients with Nasopharyngeal Carcinoma. Cancer Res. 1999, 59, 1188–1191. [Google Scholar] [PubMed]
- Sugden, D. Quantitative PCR. In Medical Biomethods Handbook; Humana Press: Totowa, NJ, USA, 2007; pp. 327–345. [Google Scholar]
- Fan, H.; Gulley, M.L. Epstein-Barr Viral Load Measurement as a Marker of EBV-Related Disease. Mol. Diagn. 2001, 6, 279–289. [Google Scholar] [CrossRef] [PubMed]
- Espy, M.J.; Uhl, J.R.; Sloan, L.M.; Buckwalter, S.P.; Jones, M.F.; Vetter, E.A.; Yao, J.D.C.; Wengenack, N.L.; Rosenblatt, J.E.; Cockerill, F.R.; et al. Real-Time PCR in Clinical Microbiology: Applications for Routine Laboratory Testing. Clin. Microbiol. Rev. 2006, 19, 165–256. [Google Scholar] [CrossRef] [PubMed]
- De Paschale, M.; Clerici, P. Serological Diagnosis of Epstein-Barr Virus Infection: Problems and Solutions. World J. Virol. 2012, 1, 31–43. [Google Scholar] [CrossRef] [PubMed]
- Iwata, S.; Shibata, Y.; Kawada, J.; Hara, S.; Nishiyama, Y.; Morishima, T.; Ihira, M.; Yoshikawa, T.; Asano, Y.; Kimura, H. Rapid Detection of Epstein–Barr Virus DNA by Loop-Mediated Isothermal Amplification Method. J. Clin. Virol. 2006, 37, 128–133. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Tang, J.; Wang, M.; Ma, Q.; Wang, Y. Visual Detection and Evaluation of Latent and Lytic Gene Expression during Epstein-Barr Virus Infection Using One-Step Reverse Transcription Loop-Mediated Isothermal Amplification. Int. J. Mol. Sci. 2013, 14, 23922–23940. [Google Scholar] [CrossRef]
- Notomi, T.; Okayama, H.; Masubuchi, H.; Yonekawa, T.; Watanabe, K.; Amino, N.; Hase, T. Loop-Mediated Isothermal Amplification of DNA. Nucleic Acids Res. 2000, 28, e63. [Google Scholar] [CrossRef]
- Elamin, A.; Ali, R.W.; Bakken, V. Putative Periodontopathic Bacteria and Herpes Viruses Interactions in the Subgingival Plaque of Patients with Aggressive Periodontitis and Healthy Controls. Clin. Exp. Dent. Res. 2017, 3, 183–190. [Google Scholar] [CrossRef]
- Bustin, S.A.; Mueller, R. Real-Time Reverse Transcription PCR (QRT-PCR) and Its Potential Use in Clinical Diagnosis. Clin. Sci. 2005, 109, 365–379. [Google Scholar] [CrossRef]
- Stein, J.M.; Yekta, S.S.; Kleines, M.; Ok, D.; Kasaj, A.; Reichert, S.; Schulz, S.; Scheithauer, S. Failure to Detect an Association between Aggressive Periodontitis and the Prevalence of Herpesviruses. J. Clin. Periodontol. 2013, 40, 1–7. [Google Scholar] [CrossRef]
- Khosropanah, H.; Karandish, M.; Ziaeyan, M.; Jamalidoust, M. Quantification of Epstein-Barr Virus and Human Cytomegalovirus in Chronic Periodontal Patients. Available online: https://sites.kowsarpub.com/jjm/articles/56432.html#abstract (accessed on 21 October 2020).
- Kato, A.; Imai, K.; Ochiai, K.; Ogata, Y. Prevalence and Quantitative Analysis of Epstein–Barr Virus DNA and Porphyromonas Gingivalis Associated with Japanese Chronic Periodontitis Patients. Clin. Oral Investig. 2015, 19, 1605–1610. [Google Scholar] [CrossRef] [PubMed]
- Passariello, C.; Gigola, P.; Testarelli, L.; Puttini, M.; Schippa, S.; Petti, S. Evaluation of Microbiota Associated with Herpesviruses in Active Sites of Generalized Aggressive Periodontitis. Ann. Stomatol. (Roma) 2017, 8, 59–70. [Google Scholar] [CrossRef] [PubMed]
- Solomon, S.M.; Filioreanu, A.M.; Stelea, C.G.; Grigoras, S.I.; Sufaru, I.G.; Maftei, G.A.; Martu, S.; Scutariu, M.M.; Popa, C. The Assessment of the Association Between Herpesviruses and Subgingival Bacterial Plaque by Real-Time PCR Analysis. Rev. Chim. 2018, 69, 507–510. [Google Scholar] [CrossRef]
- Srivastava, A.; Shukla, S.; Srivastava, P.; Dhole, T.; Nayak, M.; Nayak, A.; Mathur, A. Real Time Detection and Quantification of Epstein Barr Virus in Different Grades of Oral Gingivitis and Periodontitis Patients. J. Exp. Ther. Oncol. 2019, 13, 9–14. [Google Scholar]
- Olivieri, C.V.; Raybaud, H.; Tonoyan, L.; Abid, S.; Marsault, R.; Chevalier, M.; Doglio, A.; Vincent-Bugnas, S. Epstein-Barr Virus-Infected Plasma Cells in Periodontitis Lesions. Microb. Pathog. 2020, 143, 104128. [Google Scholar] [CrossRef]
- Gulley, M.L. Molecular Diagnosis of Epstein-Barr Virus-Related Diseases. J. Mol. Diagn. JMD 2001, 3, 1–10. [Google Scholar] [CrossRef]
- Parra, E.R.; Francisco-Cruz, A.; Wistuba, I.I. State-of-the-Art of Profiling Immune Contexture in the Era of Multiplexed Staining and Digital Analysis to Study Paraffin Tumor Tissues. Cancers 2019, 11, 247. [Google Scholar] [CrossRef]
- Nakatsuka, S.; Homma, K.; Aozasa, K. When to Use in Situ Hybridization for the Detection of Epstein-Barr Virus: A Review of Epstein-Barr Virus-Associated Lymphomas. J. Hematop. 2015, 8, 61–70. [Google Scholar] [CrossRef]
- Saboia-Dantas, C.J.; de Toledo, L.F.C.; Sampaio-Filho, H.R.; Siqueira, J.F. Herpesviruses in Asymptomatic Apical Periodontitis Lesions: An Immunohistochemical Approach. Oral Microbiol. Immunol. 2007, 22, 320–325. [Google Scholar] [CrossRef]
- Sunde, P.T.; Olsen, I.; Enersen, M.; Beiske, K.; Grinde, B. Human Cytomegalovirus and Epstein–Barr Virus in Apical and Marginal Periodontitis: A Role in Pathology? J. Med. Virol. 2008, 80, 1007–1011. [Google Scholar] [CrossRef]
- Makino, K.; Takeichi, O.; Hatori, K.; Imai, K.; Ochiai, K.; Ogiso, B. Epstein-Barr Virus Infection in Chronically Inflamed Periapical Granulomas. PLoS ONE 2015, 10, e0121548. [Google Scholar] [CrossRef] [PubMed]
- Niedobitek, G.; Herbst, H. In Situ Detection of Epstein-Barr Virus and Phenotype Determination of EBV-Infected Cells. Methods Mol. Biol. 2006, 326, 115–137. [Google Scholar] [CrossRef] [PubMed]
- Weiss, L.M.; Chen, Y.-Y. EBER in Situ Hybridization for Epstein-Barr Virus. Methods Mol. Biol. 2013, 999, 223–230. [Google Scholar] [CrossRef] [PubMed]
- Gilligan, K.; Rajadurai, P.; Resnick, L.; Raab-Traub, N. Epstein-Barr Virus Small Nuclear RNAs Are Not Expressed in Permissively Infected Cells in AIDS-Associated Leukoplakia. Proc. Natl. Acad. Sci. USA 1990, 87, 8790–8794. [Google Scholar] [CrossRef] [PubMed]
- Fok, V.; Mitton-Fry, R.M.; Grech, A.; Steitz, J.A. Multiple Domains of EBER 1, an Epstein-Barr Virus Noncoding RNA, Recruit Human Ribosomal Protein L22. RNA 2006, 12, 872–882. [Google Scholar] [CrossRef] [PubMed]
- Gulley, M.L.; Glaser, S.L.; Craig, F.E.; Borowitz, M.; Mann, R.B.; Shema, S.J.; Ambinder, R.F. Guidelines for Interpreting EBER In Situ Hybridization and LMP1 Immunohistochemical Tests for Detecting Epstein-Barr Virus in Hodgkin Lymphoma. Am. J. Clin. Pathol. 2002, 117, 259–267. [Google Scholar] [CrossRef] [PubMed]
- Wee, Y.T.F.; Alkaff, S.M.F.; Lim, J.C.T.; Loh, J.J.H.; Hilmy, M.H.; Ong, C.; Nei, W.L.; Jain, A.; Lim, A.; Takano, A.; et al. An Integrated Automated Multispectral Imaging Technique That Simultaneously Detects and Quantitates Viral RNA and Immune Cell Protein Markers in Fixed Sections from Epstein-Barr Virus-Related Tumours. Ann. Diagn. Pathol. 2018, 37, 12–19. [Google Scholar] [CrossRef]
- Ko, J.N.; Jung, J.K.; Park, Y.I.; Shin, H.J.; Huh, J.; Back, S.; Kim, Y.J.; Kim, J.H.; Go, H. Multistaining Optimization for Epstein-Barr Virus–Encoded RNA In Situ Hybridization and Immunohistochemistry of Formalin-Fixed Paraffin-Embedded Tissues Using an Automated Immunostainer. J. Pathol. Transl. Med. 2019, 53, 317–326. [Google Scholar] [CrossRef]
- Dey, P. Flow Cytometry: Basic Principles, Procedure and Applications in Pathology. In Basic and Advanced Laboratory Techniques in Histopathology and Cytology; Springer: Singapore, 2018; pp. 171–183. ISBN 978-981-10-8251-1. [Google Scholar]
- Cossarizza, A.; Chang, H.-D.; Radbruch, A.; Akdis, M.; Andrä, I.; Annunziato, F.; Bacher, P.; Barnaba, V.; Battistini, L.; Bauer, W.M.; et al. Guidelines for the Use of Flow Cytometry and Cell Sorting in Immunological Studies. Eur. J. Immunol. 2017, 47, 1584–1797. [Google Scholar] [CrossRef]
- Kimura, H.; Miyake, K.; Yamauchi, Y.; Nishiyama, K.; Iwata, S.; Iwatsuki, K.; Gotoh, K.; Kojima, S.; Ito, Y.; Nishiyama, Y. Identification of Epstein-Barr Virus (EBV)–Infected Lymphocyte Subtypes by Flow Cytometric In Situ Hybridization in EBV-Associated Lymphoproliferative Diseases. J. Infect. Dis. 2009, 200, 1078–1087. [Google Scholar] [CrossRef]
- Zachova, K.; Kosztyu, P.; Zadrazil, J.; Matousovic, K.; Vondrak, K.; Hubacek, P.; Kostovcikova, K.; Hogenova, H.T.; Mestecky, J.; Raska, M. Multiparametric Flow Cytometry Analysis of Peripheral Blood B Cell Trafficking Differences among Epstein-Barr Virus Infected and Uninfected Subpopulations. Biomed. Pap. Med. Fac. Univ. Palacky Olomouc Czechoslov. 2020, 164, 247–254. [Google Scholar] [CrossRef] [PubMed]
- Liao, X.; Makris, M.; Luo, X.M. Fluorescence-Activated Cell Sorting for Purification of Plasmacytoid Dendritic Cells from the Mouse Bone Marrow. J. Vis. Exp. JoVE 2016, e54641. [Google Scholar] [CrossRef] [PubMed]
- Contreras, A.; Zadeh, H.H.; Nowzari, H.; Slots, J. Herpesvirus Infection of Inflammatory Cells in Human Periodontitis. Oral Microbiol. Immunol. 1999, 14, 206–212. [Google Scholar] [CrossRef] [PubMed]
- Oko, L.M.; Kimball, A.K.; Kaspar, R.E.; Knox, A.N.; Coleman, C.B.; Rochford, R.; Chang, T.; Alderete, B.; van Dyk, L.F.; Clambey, E.T. Multidimensional Analysis of Gammaherpesvirus RNA Expression Reveals Unexpected Heterogeneity of Gene Expression. PLOS Pathog. 2019, 15, e1007849. [Google Scholar] [CrossRef]
- Fournier, B.; Boutboul, D.; Bruneau, J.; Miot, C.; Boulanger, C.; Malphettes, M.; Pellier, I.; Dunogué, B.; Terrier, B.; Suarez, F.; et al. Rapid Identification and Characterization of Infected Cells in Blood during Chronic Active Epstein-Barr Virus Infection. J. Exp. Med. 2020, 217, e20192262. [Google Scholar] [CrossRef]
- Wang, F.; Flanagan, J.; Su, N.; Wang, L.-C.; Bui, S.; Nielson, A.; Wu, X.; Vo, H.-T.; Ma, X.-J.; Luo, Y. RNAscope A Novel in Situ RNA Analysis Platform for Formalin-Fixed, Paraffin-Embedded Tissues. J. Mol. Diagn. 2012, 14, 22–29. [Google Scholar] [CrossRef]
- Yu, F.; Lu, Y.; Petersson, F.; Wang, D.-Y.; Loh, K.S. Presence of Lytic Epstein-Barr Virus Infection in Nasopharyngeal Carcinoma. Head Neck 2018, 40, 1515–1523. [Google Scholar] [CrossRef]
- Hildyard, J.C.W.; Rawson, F.; Wells, D.J.; Piercy, R.J. Multiplex in Situ Hybridization within a Single Transcript: RNAscope Reveals Dystrophin MRNA Dynamics. PLoS ONE 2020, 15, e0239467. [Google Scholar] [CrossRef]
- Kim, Y.; Kim, H.S.; Park, J.S.; Kim, C.J.; Kim, W.H. Identification of Epstein-Barr Virus in the Human Placenta and Its Pathologic Characteristics. J. Korean Med. Sci. 2017, 32, 1959–1966. [Google Scholar] [CrossRef]
- Freeman, W.M.; Robertson, D.J.; Vrana, K.E. Fundamentals of DNA Hybridization Arrays for Gene Expression Analysis. BioTechniques 2000, 29, 1042–1055. [Google Scholar] [CrossRef]
- Socransky, S.S.; Haffajee, A.D. Periodontal Microbial Ecology. Periodontology 2000 2005, 38, 135–187. [Google Scholar] [CrossRef] [PubMed]
- Paster, B.J.; Dewhirst, F.E. Molecular Microbial Diagnosis. Periodontology 2000 2009, 51, 38–44. [Google Scholar] [CrossRef] [PubMed]
- Debaugnies, F.; Busson, L.; Ferster, A.; Lewalle, P.; Azzi, N.; Aoun, M.; Verhaegen, G.; Mahadeb, B.; de Marchin, J.; Vandenberg, O.; et al. Detection of Herpesviridae in Whole Blood by Multiplex PCR DNA-Based Microarray Analysis after Hematopoietic Stem Cell Transplantation. J. Clin. Microbiol. 2014, 52, 2552–2556. [Google Scholar] [CrossRef] [PubMed]
- Farisyi, M.A.; Sufiawati, I. Detection of Epstein–Barr Virus DNA in Saliva of HIV-1-Infected Individuals with Oral Hairy Leukoplakia. Oral Dis. 2020, 26, 158–160. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Zhou, P.; Cao, F.; Lin, Z.; Liang, D.; Zhou, X. Expression Profiling and Cell Type Classification Analysis in Periodontitis Reveal Dysregulation of Multiple LncRNAs in Plasma Cells. Front. Genet. 2020, 11, 382. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Fang, X.; Feng, Z.; Guo, Y.-M.; Peng, R.-J.; Liu, T.; Huang, Z.; Feng, Y.; Sun, X.; Xiong, Z.; et al. Direct Sequencing and Characterization of a Clinical Isolate of Epstein-Barr Virus from Nasopharyngeal Carcinoma Tissue by Using Next-Generation Sequencing Technology. J. Virol. 2011, 85, 11291–11299. [Google Scholar] [CrossRef]
- Shendure, J.; Ji, H. Next-Generation DNA Sequencing. Nat. Biotechnol. 2008, 26, 1135–1145. [Google Scholar] [CrossRef]
- Ansorge, W.J. Next-Generation DNA Sequencing Techniques. New Biotechnol. 2009, 25, 195–203. [Google Scholar] [CrossRef]
- Miller, R.R.; Montoya, V.; Gardy, J.L.; Patrick, D.M.; Tang, P. Metagenomics for Pathogen Detection in Public Health. Genome Med. 2013, 5, 81. [Google Scholar] [CrossRef]
- Di Resta, C.; Galbiati, S.; Carrera, P.; Ferrari, M. Next-Generation Sequencing Approach for the Diagnosis of Human Diseases: Open Challenges and New Opportunities. EJIFCC 2018, 29, 4–14. [Google Scholar]
- Lin, Z.; Xu, G.; Deng, N.; Taylor, C.; Zhu, D.; Flemington, E.K. Quantitative and Qualitative RNA-Seq-Based Evaluation of Epstein-Barr Virus Transcription in Type I Latency Burkitt’s Lymphoma Cells. J. Virol. 2010, 84, 13053–13058. [Google Scholar] [CrossRef] [PubMed]
- Strong, M.J.; Xu, G.; Coco, J.; Baribault, C.; Vinay, D.S.; Lacey, M.R.; Strong, A.L.; Lehman, T.A.; Seddon, M.B.; Lin, Z.; et al. Differences in Gastric Carcinoma Microenvironment Stratify According to EBV Infection Intensity: Implications for Possible Immune Adjuvant Therapy. PLoS Pathog. 2013, 9, e1003341. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.; Lin, Z.; Wu, Y.; Dong, J.; Zhao, B.; Cheng, Y.; Huang, P.; Xu, L.; Xia, T.; Xiong, D.; et al. Comprehensive Profiling of EBV Gene Expression in Nasopharyngeal Carcinoma through Paired-End Transcriptome Sequencing. Front. Med. 2016, 10, 61–75. [Google Scholar] [CrossRef]
- Kim, Y.-G.; Kim, M.; Kang, J.H.; Kim, H.J.; Park, J.-W.; Lee, J.-M.; Suh, J.-Y.; Kim, J.-Y.; Lee, J.-H.; Lee, Y. Transcriptome Sequencing of Gingival Biopsies from Chronic Periodontitis Patients Reveals Novel Gene Expression and Splicing Patterns. Hum. Genomics 2016, 10, 28. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: A Revolutionary Tool for Transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef] [PubMed]