The Eph receptor tyrosine kinase family is activated by binding to their cognate ephrin ligands and represents important components of signalling pathways involved in animal development.
- Eph receptors
- EphA2
- ephrin
1. Introduction
The Eph family of receptor tyrosine kinases (RTKs), involved in signalling pathways that are key to embryogenesis and tissue patterning, have been implicated in the oncogenesis of a number of cancers. Generally, this involves their aberrant expression, allowing them to act as either tumour promoters or tumour suppressors, depending on the context[1][2]. Eph receptors have been described to play a role in breast cancer, colorectal cancer, lung cancer, prostate cancer and brain cancer[3]. In addition, more recent evidence for the involvement of Eph receptors in Kaposi’s sarcoma (KS)[4][5], the most common acquired immune deficiency syndrome (AIDS)-related malignancy worldwide, has been investigated[6]. Understanding the oncogenic mechanisms of Eph receptors, however, proves to be a challenge due to the fact that both canonical and noncanonical pathways exist. The most well-characterised example of this is for EphA2, where the classical ligand- and tyrosine kinase-dependent signalling mechanism is accompanied by a pathway in which tumour promotion is achieved independently of ligand or tyrosine kinase activation of the receptor[7].
2. Eph Receptor Structure and Signalling
Eph receptors are type-I transmembrane proteins with a structure that is generally conserved. The ligand-binding domain, cysteine-rich region and two fibronectin type III repeats compose the extracellular domain of the receptor, while the intracellular region is made up of a juxtamembrane domain, a protein tyrosine kinase (Pkinase-Tyr) domain, a sterile alpha motif (SAM) and a C-terminal PDZ-binding motif (Figure 1)[2][8]. There are two classes of Eph receptors, grouped according to the ligands they preferentially bind. While there are a few exceptions, EphA-type receptors bind ephrin-A ligands and the EphB-type receptors bind ephrin-B ligands (Figure 1)[8]. The ephrin ligands are generally membrane-bound, and it is the difference in anchorage that distinguishes the two classes. Ephrin-A ligands are attached to the membrane via a glycophosphatidylinositol anchor; this is in contrast to the ephrin-B ligands, which have a transmembrane domain, as well as a cytoplasmic region with a PDZ domain[8][9]. Heterodimerisation upon interaction between an Eph receptor and its ephrin ligand is followed by the formation of tetrameric complexes, leading to receptor tyrosine phosphorylation and kinase activation[9]. A unique feature of Eph-ephrin signalling is that it is bidirectional. Conventional forward signalling is that already mentioned, in which the signal is transduced in the receptor-expressing cell. Reverse signalling, on the other hand, involves a signal transduction cascade in the ephrin-expressing cell. For example, upon Eph receptor engagement, the cytoplasmic tail of the ephrin-B ligand becomes tyrosine phosphorylated and can then interact with signalling molecules that contain SRC-homology-2 domains [8][9][10].
This signalling plays a role in a number of biological processes important for both development and homeostasis. By modifying cell adhesion and the organisation of the actin cytoskeleton, Eph signalling controls cell morphology and migration. Eph signalling also affects cell proliferation and differentiation[10][11]. Many of these functions are also important in cancer development, when well-controlled functions become dysregulated. Hence, in addition to their expression in normal tissues, Eph receptors are expressed in cancer cells and the tumour microenvironment where they are involved in processes related to tumorigenesis and metastasis[9][11][12]. Expression in tumours, however, is not always increased, and the downregulation of certain Eph molecules in a number of malignancies suggests that Eph receptors can act as both tumour promoters and suppressors[2][11].

Figure 1. The general structure of the Eph receptors and ephrin ligands. Both the ephrin-A and ephrin-B ligands are depicted here. Figure created with BioRender.com.
3. EphA2 and Oncogenesis
EphA2, for example, plays an important role in a number of cancers[3]; however, its role is context-dependent, and it can act as either a tumour promoter or tumour suppressor. There has been an accumulation of evidence that shows that EphA2 possesses peculiar modes of signalling and it may be that these underscore its opposing functions[7]. EphA2 has both canonical and non-canonical modes of driving oncogenesis (Figure 2). The canonical pathway involves ligand-and tyrosine kinase-dependent forward signalling via EphA2, which suppresses tumorigenesis. It does so through the inhibition of FAK, Akt and ERK phosphorylation, thereby controlling cell motility and survival[7]. For example, upon EphA2 autophosphorylation, it can no longer associate with FAK to cause phosphorylation and activation. FAK has been implicated in the growth of breast cancer cell lines and its deactivation resulted in reduced oncogenic activity[13]. Importantly, this mechanism is specifically reliant onTyr772 phosphorylation, highlighted by the fact that a phosphorylation-abrogating Tyr772 mutation resulted in increased transendothelial migration[14]. Consequently, a low level of EphA2 forward signalling promotes tumorigenicity. In the context of breast cancer, this could be due to low ephrin expression, an impaired EphA2-ephrin-A1 interaction due to loss of cadherin, or dephosphorylation of Tyr772 by LMW-PTP[11][15][16]. A good example of this duality has been identified with regards to mesothelioma. Here, EphA2 activation by ephrin-A1 is associated with suppressed tumorigenesis. However, in mesothelioma cell lines, EphA2 was found to be overexpressed, and therefore, in the absence of sufficient ligand, the signalling of other RTKs through the Ras oncogene results in the promotion of malignancy[17].
The noncanonical pathway, conversely, involves the ligand- and tyrosine kinase-independent activation and phosphorylation of EphA2. This is regulated by inflammatory cytokines and growth factors via phosphorylation of EphA2 at Ser897, with induction of this phosphorylation carried out by RSK, Akt and protein kinase A (PKA)[7]. The effects of this phosphorylation include localisation of EphA2 at the leading edge of migrating cells, allowing for actin cytoskeleton assembly and the formation of lamellipodia[18], thereby leading to the promotion and maintenance of certain cancer cell features such as motility and proliferation[7]. A recent demonstration of the noncanonical EphA2 action was focused on its role in the oncogenesis of bladder cancer. Here, the growth factor progranulin was found to be the predominant EphA2 ligand and was upregulated compared to ephrin-A1, which was expressed at normal levels. Stimulation of EphA2 by progranulin resulted in Ser897 phosphorylation, allowing for interaction with liprin-α1, a protein that is necessary for cell motility to occur in this case[19]. 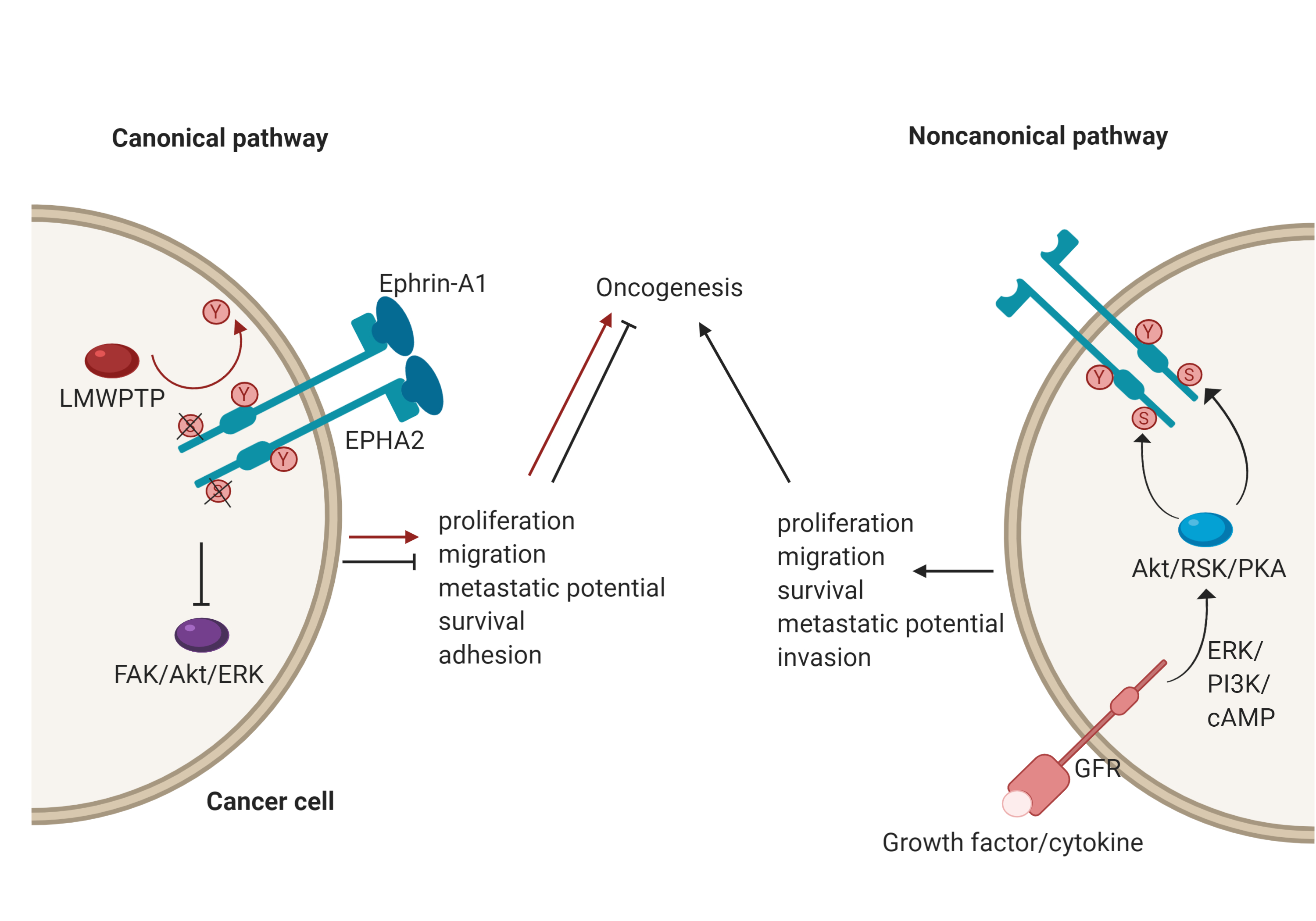
References
- Maria Genander; Jonas Frisén; Ephrins and Eph receptors in stem cells and cancer. Current Opinion in Cell Biology 2010, 22, 611-616, 10.1016/j.ceb.2010.08.005.
- Chung-Ting Jimmy Kou; Raj P. Kandpal; Differential Expression Patterns of Eph Receptors and Ephrin Ligands in Human Cancers. BioMed Research International 2018, 2018, 1-23, 10.1155/2018/7390104.
- Meg Anderton; Emma Van Der Meulen; Melissa J. Blumenthal; Georgia Schäfer; The Role of the Eph Receptor Family in Tumorigenesis. Cancers 2021, 13, 206, 10.3390/cancers13020206.
- Melissa J. Blumenthal; Charlotte Schutz; Graeme Meintjes; Zainab Mohamed; Marc Mendelson; Jon M. Ambler; Denise Whitby; Romel D. Mackelprang; Sinead Carse; Arieh A. Katz; et al.Georgia Schäfer EPHA2 sequence variants are associated with susceptibility to Kaposi's sarcoma-associated herpesvirus infection and Kaposi's sarcoma prevalence in HIV-infected patients.. Cancer Epidemiology 2018, 56, 133-139, 10.1016/j.canep.2018.08.005.
- Melissa J. Blumenthal; Elena Maria Cornejo Castro; Denise Whitby; Arieh A. Katz; Georgia Schäfer; Evidence for altered host genetic factors in KSHV infection and KSHV ‐related disease development. Reviews in Medical Virology 2020, e2160, 1-13, 10.1002/rmv.2160.
- Enrique A. Mesri; Ethel Cesarman; Chris Boshoff; Kaposi's sarcoma and its associated herpesvirus. Nature Cancer 2010, 10, 707-719, 10.1038/nrc2888.
- Yue Zhou; Hiroaki Sakurai; Emerging and Diverse Functions of the EphA2 Noncanonical Pathway in Cancer Progression. Biological & Pharmaceutical Bulletin 2017, 40, 1616-1624, 10.1248/bpb.b17-00446.
- Hanna Surawska; Patrick C. Ma; Ravi Salgia; The role of ephrins and Eph receptors in cancer. Cytokine & Growth Factor Reviews 2004, 15, 419-433, 10.1016/j.cytogfr.2004.09.002.
- Julio Castaño; Veronica Davalos; Simo Schwartz Jr; Diego Arango; EPH receptors in cancer. Histololgy and Histopathology 2008, 23(8), 1011-23, 10.14670/HH-23.1011.
- Elena B. Pasquale; Eph receptor signalling casts a wide net on cell behaviour. Nature Reviews Molecular Cell Biology 2005, 6, 462-475, 10.1038/nrm1662.
- Elena B. Pasquale; Eph receptors and ephrins in cancer: bidirectional signalling and beyond. Nature Cancer 2010, 10, 165-180, 10.1038/nrc2806.
- Elena B. Pasquale; Eph-Ephrin Bidirectional Signaling in Physiology and Disease. Cell 2008, 133, 38-52, 10.1016/j.cell.2008.03.011.
- Masaru Nakamoto; Andrew D. Bergemann; Diverse roles for the Eph family of receptor tyrosine kinases in carcinogenesis. Microscopy Research and Technique 2002, 59, 58-67, 10.1002/jemt.10177.
- Marie Locard-Paulet; Lindsay Lim; Giulia Veluscek; Kelly M McMahon; John Sinclair; Antoinette Van Weverwijk; Jonathan D. Worboys; Yinyin Yuan; Clare M. Isacke; Claus Jørgensen; et al. Phosphoproteomic analysis of interacting tumor and endothelial cells identifies regulatory mechanisms of transendothelial migration. Science Signaling 2016, 9, ra15-ra15, 10.1126/scisignal.aac5820.
- Keith D. Kikawa; Derika R. Vidale; Robert L. Van Etten; Michael S. Kinch; Regulation of the EphA2 Kinase by the Low Molecular Weight Tyrosine Phosphatase Induces Transformation. Journal of Biological Chemistry 2002, 277, 39274-39279, 10.1074/jbc.m207127200.
- N D Zantek; M Azimi; M Fedor-Chaiken; B Wang; R Brackenbury; Michael S. Kinch; E-cadherin regulates the function of the EphA2 receptor tyrosine kinase.. Cell growth & differentiation : the molecular biology journal of the American Association for Cancer Research 1999, 10, 629–638.
- Najmunnisa Nasreen; Nazli Khodayari; Kamal A Mohammed; Advances in malignant pleural mesothelioma therapy: targeting EphA2 a novel approach. American journal of cancer research 2012, 2, 222-234.
- Hui Miao; Da-Qiang Li; Amitava Mukherjee; Hong Guo; Aaron Petty; Jennifer Cutter; James P. Basilion; John Sedor; Jiong Wu; David Danielpour; et al.Andrew E. SloanMark L. CohenBingcheng Wang EphA2 Mediates Ligand-Dependent Inhibition and Ligand-Independent Promotion of Cell Migration and Invasion via a Reciprocal Regulatory Loop with Akt. Cancer Cell 2009, 16, 9-20, 10.1016/j.ccr.2009.04.009.
- Simone Buraschi; Thomas Neill; Shi-Qiong Xu; Chiara Palladino; Antonino Belfiore; Renato V. Iozzo; Andrea Morrione; Progranulin/EphA2 axis: A novel oncogenic mechanism in bladder cancer. Matrix Biology 2020, 93, 10-24, 10.1016/j.matbio.2020.03.009.