Protein degradation maintains cellular integrity by regulating virtually all biological pro- cesses, whereas impaired proteolysis perturbs protein quality control, and often leads to human disease. Two major proteolytic systems are responsible for protein breakdown in all cells: autophagy, which facilitates the loss of organelles, protein aggregates, and cell surface proteins; and the ubiquitin-proteasome system (UPS), which promotes degradation of mainly soluble proteins. Recent findings indicate that more complex protein structures, such as filamentous assemblies, which are not acces- sible to the catalytic core of the proteasome in vitro, can be efficiently degraded by this proteolytic machinery in systemic catabolic states in vivo. Mechanisms that loosen the filamentous structure seem to be activated first, hence increasing the accessibility of protein constituents to the UPS. In this review, we will discuss the mechanisms underlying the disassembly and loss of the intricate insoluble filamentous myofibrils, which are responsible for muscle contraction, and whose degradation by the UPS causes weakness and disability in aging and disease. Several lines of evidence indicate that myofibril breakdown occurs in a strictly ordered and controlled manner, and the function of AAA-ATPases is crucial for their disassembly and loss.
- ubiquitin
- proteasome
- autophagy
- muscle atrophy
- intermediate filaments
- myofibrils
- AAA-ATPases
1. Introduction
Proteolysis promotes tissue homeostasis by controlling protein abundance in response to extracellular and intracellular cues, and by preventing accumulation of misfolded or damaged proteins [1]. Conversely, unbalanced protein breakdown can lead to tissue wasting, accumulation of abnormal proteins, and disease (e.g., neurodegenerative diseases such as Alzheimer’s, Parkinson’s, and Huntington disease [2]). The major proteolytic system responsible for degradation of soluble proteins is the ubiquitin-proteasome system (UPS) [3], while autophagy generally facilitates the loss of organelles, and cell surface or aggregated proteins. The vast majority of soluble proteins destined for degradation by the proteasome are initially tagged by the covalent attachment of ubiquitin moieties [4][5][4,5]. In addition, intricate filamentous structures, such as the myofibrillar apparatus in striated muscles, are also ubiquitinated and efficiently degraded by the UPS in vivo, although they seem resistant to this proteolytic machinery in vitro [6]. The mechanism for myofibril breakdown has long been a mystery, and recent evidence indicates that a tightly regulated solubilization process is required for efficient degradation. The loss of this fundamental contractile machinery involves an initial degradation of the cytoskeletal networks and structural and regulatory proteins that stabilize the myofibril.
2. Myofibrils are an Intricate Filamentous Structure
Skeletal muscles support the skeleton and promote motion by contraction. They are composed of a bundle of long multinuclear cells, whose volume is mainly occupied by a precisely aligned filament system of myofibrils that is responsible for force production [7]. This contractile machinery is organized in repeated units of sarcomeres, which are delimited by Z-bands, and mainly contain myosin in thick, and actin in thin, filaments (Figure 1). The Myosin molecule is composed of two heavy chains (MyHC), and two pairs of regulatory (MyLC2) and essential (MyLC1) light chains, and is divided into three main regions: (1) the N-terminal head domain, which contains the motor domain that binds actin and hydrolyzes ATP to generate force; (2) the C-terminal α-helical tail domain, that constitutes the backbone of the thick filaments; and (3) the neck domain, which links the head and tail domains, and represents the site where MyLCs non-covalently bind MyHC [8]. Myosin thick filaments are stabilized at the center of the sarcomere by additional structural and regulatory proteins. For example, MyLCs, and myosin-binding protein C (MyBP-C), which binds the thick filament periodically, are required for myofibril stability and normal contractility [9][10][11][9,10,11]. In addition, ubiquitin ligases, proteases (e.g., calpain-3), and kinases bind myosin and regulate its activity [12].
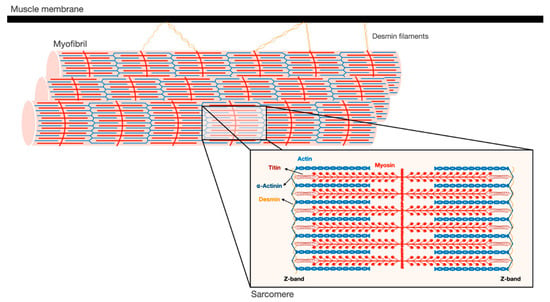
Figure 1. Schematic illustration of the myofibrillar apparatus. Myofibrils are a highly ordered filament system, organized in repeating units of sarcomeres. The Z-bands are the boundaries of the sarcomere and the sites where the actin thin filaments and desmin intermediate filaments are bound. Myosin thick filaments span the center of the sarcomere, and slide over actin filaments toward the Z-bands when the muscle contracts. These filaments are stabilized by additional structural proteins, such as α-actinin, which cross-links actin and desmin at the Z-bands. In addition, desmin intermediate filaments contribute to muscle architecture and contractile function by linking adjacent myofibrils laterally, and to the muscle membrane and cellular organelles.
The Z-bands constitute the anchoring site for actin thin filaments, as well as for two other filament systems in the myofibril, titin and nebulin. The thin filaments form a helical arrangement, and the protein tropomyosin lies in the α-helical grooves that are formed along the filaments. Aligned at intervals along the actin filaments is the protein complex, troponin (Tn), which is composed of three components Tn-T, Tn-I, and Tn-C, and represents the on-off switch of muscle contraction [7]. Muscle contraction is activated by Ca2+ influx into the cytosol, which in turn binds Tn-C, causing displacement of Tn-T and Tn-I from tropomyosin, and consequently leading to exposure of myosin binding sites on actin. Then, myosin heads bind actin and move toward the Z-bands by converting the chemical energy produced by ATP hydrolysis into mechanical force. The sliding of the thick over the thin filaments in their overlapping regions causes shortening of the sarcomeres and the muscle fiber, and powers contraction.
At the Z-bands, the thin filaments bind the predominant intermediate filament (IF) protein in muscle, desmin, via cross-linking proteins, such as α-actinin (Figure 1). Desmin IF contribute to the mechanical and structural integrity of myofibrils by linking adjacent myofibrils laterally, and to the sarcolemma, nucleus, and mitochondria, and by promoting muscle homeostasis [13][14][15][13,14,15]. Loss of desmin IF during atrophy induced by fasting or denervation (i.e., loss of motor nerve input) precedes and promotes myofibril destruction [16][17][18][16,17,18]. Therefore, the loss of structural or regulatory components that contribute to myofibril stability (e.g., MyLCs, MyBP-C, desmin filaments) is likely to be an early event leading to myofibril breakdown.
3. Ubiquitin Ligases Can Act on Insoluble Filaments
The highly organized and complex structure of myofibrils is degraded upon fasting, denervation, and aging, and in many human diseases (e.g., cancer [19][20][19,20], diabetes [21][22][21,22], sepsis [23][24][23,24], and renal [25] and cardiac failure [26]), leading to loss of muscle mass and strength (i.e., atrophy). As muscles continue to contract in these various catabolic states, myofibril breakdown by the UPS must occur in a highly ordered and tightly regulated manner. Two muscle specific ubiquitin ligases, Muscle RING finger 1 (MuRF1) and Atrogin-1/MAFbx are induced in most types of atrophy, and promote proteolysis [27][28][27,28], while deficiency of either enzyme attenuates wasting [28][29][28,29]. During atrophy, Atrogin-1/MAFbx catalyzes the degradation of proteins that promote protein synthesis, whereas other ligases, in particular MuRF1 and ubiquitin tripartite motif-containing protein 32 (Trim32), catalyze the ubiquitination of myofibrillar proteins (i.e., myosin, actin, and associated proteins) and the desmin cytoskeleton [16][17][18][30][31][32][16,17,18,30,31,32]. Although ubiquitin ligases normally act on soluble substrates, recent studies surprisingly demonstrated that they can also catalyze the ubiquitination of proteins within the insoluble myofibril.
In vitro studies in muscle extracts have demonstrated that purified actin and myosin can be efficiently degraded by the proteasome [6][33][6,33], but not when these proteins are associated with each other in the actomyosin complex or intact myofibrils. These findings suggested that the UPS may not, by itself, be able to degrade components of intact myofibrils, and constituent proteins must be released by some mechanism to be substrates for degradation [6]. Consequently, several groups have suggested that proteases, such as calpains or caspases, may cleave the myofibril to accelerate disassembly before ubiquitination [34][35][36][34,35,36]. While this could be a possible mechanism, it has not been proven in atrophying muscles in vivo. In addition, in atrophying mouse muscles, proteolytic cleavage by calpains facilitates desmin filament loss, which is linked to myofibril destruction [17][37][38][17,37,38], but is not required for ubiquitination of the myofibrillar apparatus [30]. Cohen and colleagues showed that the monomeric RING E3s, MuRF1, and Trim32, are able to act directly on cytoskeletal networks and the insoluble myofibrils in vitro [16][30][16,30]. MyBP-C and MyLC2, which are important for thick filament stability, can be efficiently ubiquitinated by MuRF1 in vitro when still associated with the myofibril, and their selective loss in atrophying denervated mouse muscles in vivo precedes degradation of the major thick filament component, MyHC. Similarly, components of thin filament and Z-bands, including actin, tropomyosin, and α-actinin in intact myofibrils, as well as the desmin cytoskeleton are efficiently ubiquitinated by recombinant Trim32 [16][30][16,30], and degradation of desmin filaments during fasting or denervation precedes and promotes myofibril destruction. These findings suggest that ubiquitin ligases can ubiquitinate the proteins of intact myofibrils, and that the selective initial loss of proteins that stabilize the myofibril (such as MyLCs, MyBP-C, and desmin IF) may loosen this structure, rendering its protein constituents more susceptible for the catalytic activity of proteolytic enzymes and the proteasome. Accordingly, myofibrils isolated from 14-day denervated muscles, when myofibrillar myosin and actin are rapidly ubiquitinated and degraded [30], were more efficiently ubiquitinated by Trim32 in vitro than myofibrils from 10-day denervated muscles, in which degradation of myosin and actin was slow [18]. Thus, when desmin filaments are solubilized and protein ubiquitination is accelerated, myofibrillar constituents are more sensitive to Trim32, and probably to other ubiquitin ligases. Trim32 and certain deubiquitinating enzymes (including USP1 [39] and USP19 [40]) also promote myofibril breakdown indirectly, i.e., by reducing the major growth pathway in muscle, PI3K-Akt-FoxO signaling [41][42][41,42], resulting in increased proteolysis and decreased protein synthesis [43].
As mentioned above, in addition to its role in promoting thin filament loss, Trim32 is also responsible for the ubiquitination and the resulting degradation of desmin IF. Desmin filament phosphorylation by the protein kinase GSK3β [16][17][44][16,17,44] is required to enhance ubiquitination by Trim32; however, phosphorylation of Trim32′s substrates cannot be a prerequisite for ubiquitination because this enzyme can also ubiquitinate pure non-phosphorylated actin in vitro [16]. In addition, protein phosphorylation can rather protect from degradation, by preventing ubiquitination. For instance, phosphorylation of p53 upon DNA damage stabilizes this protein by reducing its association with the ubiquitin ligase, Mdm2 [45]. It has long been known that SCF ubiquitin ligases act preferentially on phosphorylated substrates (e.g., Cyclin D1, Cyclin E, β -Catenin) [5][46][47][48][5,46,47,48], a property that was considered unusual for a RING E3 such as Trim32. However, in recent years other RING E3s, in addition to Trim32, have been shown to act preferentially on phosphorylated substrates, including c-Cbl and C-terminal Hsp70-interacting protein (CHIP) [49][50][49,50].
4. Loss of Stabilizing Structures is a Prerequisite to Myofibril Breakdown
Desmin is a cytoskeletal type III IF protein (53 kDa) consisting of a N-terminal head, a central α-helical rod, and a C-terminal tail domains [51]. It is specifically expressed in muscle, and is critical for maintenance of tissue architecture and function. While desmin null mice are viable, they exhibit loss of lateral alignment of myofibrils and abnormal mitochondrial organization in both skeletal and cardiac muscles [52]. Mutations in desmin cause a variety of skeletal and cardiac myopathies, called desminopathies, which are characterized by toxic aggregation of mutant misfolded desmin. Consequently, myofibril function is perturbed, as well as mitochondrial homeostasis [53], leading to muscle weakness, respiratory dysfunction, and cardiomyopathies [54]. Since desmin IF are important for sarcomere stability and alignment, their initial loss during atrophy represents a key step in the process of myofibril instability and ultimate breakdown.
The sequence of cellular events leading to desmin filament loss has been recently discovered in studies on denervated mouse muscles. Early after denervation (3 days), desmin filaments are phosphorylated by the kinase GSK3-β, ubiquitinated by Trim32 (and probably other ubiquitin ligases), and then depolymerized by the Ca2+-dependent protease calpain-1 (Figure 2) [17][55][17,55]. These events precede significant ubiquitination and degradation of myofibrillar proteins, suggesting that the dissociation and loss of desmin filaments precede, and thus may promote, myofibril breakdown (Figure 2). In fact, introducing a dominant-negative mutant of desmin, which promotes desmin disassembly, accelerated myofibril destruction in mouse denervated muscles, indicating that loss of desmin filaments is linked to degradation of myofibrillar proteins [18]. Although desmin IF loss appears important for myofibril solubilization and proteasomal degradation, it is dispensable for myofibril ubiquitination. Accordingly, inhibition of desmin filament loss (e.g., by GSK3-β inhibition, or by downregulation of Trim32 or calpain-1) resulted in accumulation of ubiquitinated intact myofibrils in the insoluble fraction [16][17][16,17].
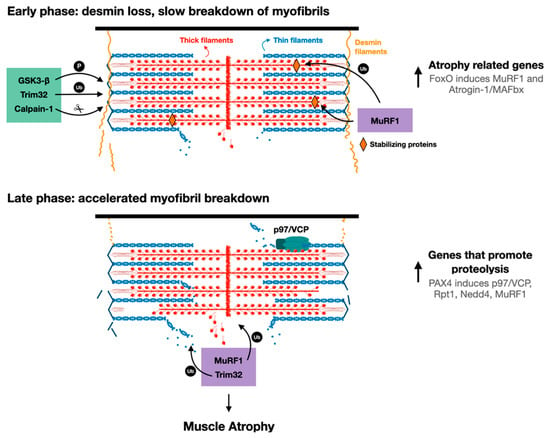
Figure 2. Proposed model for myofibril breakdown in atrophy. The sequence of events leading to myofibril breakdown and atrophy seems to define two major phases. In the early phase, there is a selective loss of desmin filaments and other proteins that stabilize the myofibrils, and the myofibrils are only slowly degraded. This initial loss of stabilizing systems accelerates myofibril breakdown in a more delayed phase by enzymes that promote proteolysis (e.g., UPS components such as E3s, p97/VCP), whose expression is increased by PAX4.
In denervated mouse muscles, increased phosphorylation of desmin filaments facilitated ubiquitination, but was insufficient to cause depolymerization, which occurred only 4 days later. This delay in desmin filament dissociation and loss was intriguing, and suggested that additional signals beyond phosphorylation and ubiquitination are required to promote desmin filament depolymerization and loss. Accordingly, loss of desmin filaments occurred at 7 days after denervation when cytosolic Ca2+ levels rose and activated calpain-1 [17]. Therefore, the timing of the elevation in Ca2+ levels may be rate limiting for desmin filament loss and myofibril destruction. Consistent with this idea, Ca2+ levels rise in different types of atrophy (e.g., fasting [17], sepsis [56][57][56,57], and cancer [58]) and in diseased muscles. For example, in mdx mice model for Duchenne muscular dystrophy, Ca2+ levels are elevated and induce protein degradation, most likely by stimulating Ca2+-specific proteases [59][60][59,60].
These investigations indicated that calpain-1 promotes the rapid dissociation of phosphorylated and ubiquitinated desmin filaments. A time-course analysis of desmin cleavage by calpain-1 in vitro demonstrated that desmin filaments, which were pretreated with the alkaline protein phosphatase calf intestinal (CIP), were less sensitive to cleavage by calpain-1. In addition, in atrophying muscles expressing GSK3-β dominant-negative inhibitor (a kinase-dead mutant), desmin filaments accumulated in their unmodified form, and were less efficiently cleaved by calpain-1 in vitro. Thus, calpain-1 shows a preference for phosphorylated desmin filaments [17]. The discovery that in atrophying muscles, phosphorylated and ubiquitinated desmin filaments are cleaved by calpain-1 is surprising because calpain-1 does not harbor a bona fide ubiquitin binding domain. Possibly, ubiquitination of desmin filaments facilitates depolymerization by triggering conformational changes that expose calpain-1 cleavage sites on desmin. Accordingly, two ubiquitination sites have been identified in the desmin rod domain [61], the only structured region in desmin monomers. In addition, it has been previously suggested that ubiquitination can cause conformational changes that expose calpain cleavage sites on substrate proteins [62][63][62,63]: Watkins et al. demonstrated that calpain-mediated cleavage of the regulatory domain of protein phosphatase 2A (PP2A) occurs after PP2A monoubiquitination by the ubiquitin ligase Midline-1, leading to PP2A degradation by the proteasome [55]. A similar role for ubiquitination has been demonstrated for interleukin-1 receptor, type 1 (IL1-R1): overexpression of IL1-R1 and its ubiquitin ligase, tumor necrosis factor receptor-associated factor-6 (TRAF6), in HEK293T cells led to IL1-R1 ubiquitination by TRAF6, which facilitated IL1-R1 cleavage by γ-secretase and loss [64]. Based on these findings, the authors suggested that ubiquitination-mediated cleavage could be a molecular mechanism controlling protein function and/or stability. Desmin filaments are polyubiquitinated by Trim32 in atrophying mouse muscles, and also in vitro by recombinant Trim32, and inhibition of their ubiquitination (e.g., by Trim32 downregulation) prevents depolymerization and atrophy [16][17][16,17]. This ubiquitination can also promote desmin filament depolymerization by facilitating binding of AAA-ATPase complexes, which extract ubiquitinated proteins from larger structures through ATP hydrolysis [65], and thus enhance their accessibility to the catalytic core of the proteasome. For example, the AAA-ATPase, p97/VCP, promotes myofibril disassembly during atrophy by facilitating the release of ubiquitinated myofibrillar proteins into the cytosol (see below) [18][66][18,66]. Its role (and the role of other AAA-ATPases) in promoting desmin loss in atrophy has not been investigated, and is an important question for future research.
Reference (Editors will rearrange the references after the entry is submitted)
- Goldberg, A.L. Protein degradation and protection against misfolded or damaged proteins. Nat. Cell Biol. 2003, 426, 895–899.
- Soto, C.; Estrada, L.D. Protein Misfolding and Neurodegeneration. Arch. Neurol. 2008, 65, 184–189.
- Lecker, S.H.; Goldberg, A.L.; Mitch, W.E. Protein Degradation by the Ubiquitin–Proteasome Pathway in Normal and Disease States. J. Am. Soc. Nephrol. 2006, 17, 1807–1819.
- Lu, K.; Brave, F.D.; Jentsch, S. Pathway choice between proteasomal and autophagic degradation. Autophagy 2017, 13, 1799–1800.
- Glickman, M.H.; Ciechanover, A. The Ubiquitin-Proteasome Proteolytic Pathway: Destruction for the Sake of Construction. Physiol. Rev. 2002, 82, 373–428.
- Solomon, V.; Goldberg, A.L. Importance of the ATP-Ubiquitin-Proteasome Pathway in the Degradation of Soluble and Myofibrillar Proteins in Rabbit Muscle Extracts. J. Biol. Chem. 1996, 271, 26690–26697.
- Rogers, D. Skeletal Muscle Structure, Function and Plasticity. Physiotherapy 2003, 89, 565.
- Aguilar, H.N.; Mitchell, B.F. Physiological pathways and molecular mechanisms regulating uterine contractility. Hum. Reprod. Updat. 2010, 16, 725–744.
- Chen, Z.; Huang, W.; Dahme, T.; Rottbauer, W.; Ackerman, M.J.; Xu, X. Depletion of zebrafish essential and regulatory myosin light chains reduces cardiac function through distinct mechanisms. Cardiovasc. Res. 2008, 79, 97–108.
- Rottbauer, W.; Wessels, G.; Dahme, T.; Just, S.; Trano, N.; Hassel, D.; Burns, C.G.; Katus, H.A.; Fishman, M.C. Cardiac Myosin Light Chain-2. Circ. Res. 2006, 99, 323–331.
- Yang, Q.; Sanbe, A.; Osinska, H.; E Hewett, T.; Klevitsky, R.; Robbins, J. A mouse model of myosin binding protein C human familial hypertrophic cardiomyopathy. J. Clin. Investig. 1998, 102, 1292–1300.
- Clark, K.A.; McElhinny, A.S.; Beckerle, M.C.; Gregorio, C.C. Striated Muscle Cytoarchitecture: An Intricate Web of Form and Function. Annu. Rev. Cell Dev. Biol. 2002, 18, 637–706.
- Mendez, M.G.; Kojima, S.; Goldman, R.D. Vimentin induces changes in cell shape, motility, and adhesion during the epithelial to mesenchymal transition. FASEB J. 2010, 24, 1838–1851.
- Chou, Y.-H.; Kuo, W.-L.; Rosner, M.R.; Tang, W.-J.; Goldman, R.D. Structural changes in intermediate filament networks alter the activity of insulin-degrading enzyme. FASEB J. 2009, 23, 3734–3742.
- Dechat, T.; Adam, S.A.; Goldman, R.D. Nuclear lamins and chromatin: When structure meets function. Adv. Enzym. Regul. 2009, 49, 157–166.
- Cohen, S.; Zhai, B.; Gygi, S.P.; Goldberg, A.L. Ubiquitylation by Trim32 causes coupled loss of desmin, Z-bands, and thin filaments in muscle atrophy. J. Cell Biol. 2012, 198, 575–589.
- Aweida, D.; Rudesky, I.; Volodin, A.; Shimko, E.; Cohen, S. GSK3-β promotes calpain-1–mediated desmin filament depolymerization and myofibril loss in atrophy. J. Cell Biol. 2018, 217, 3698–3714.
- Volodin, A.; Kosti, I.; Goldberg, A.L.; Cohen, S. Myofibril breakdown during atrophy is a delayed response requiring the transcription factor PAX4 and desmin depolymerization. Proc. Natl. Acad. Sci. USA 2017, 114, E1375–E1384.
- Lazarus, D.D.; Destree, A.T.; Mazzola, L.M.; McCormack, T.A.; Dick, L.R.; Xu, B.; Huang, J.Q.; Pierce, J.W.; Read, M.A.; Coggins, M.B.; et al. A new model of cancer cachexia: Contribution of the ubiquitin-proteasome pathway. Am. J. Physiol. Content 1999, 277, E332–E341.
- Llovera, M.; García-Martínez, C.; Agell, N.; Marzábal, M.; López-Soriano, F.J.; Argilés, J.M. Ubiquitin gene expression is increased in skeletal muscle of tumour-bearing rats. FEBS Lett. 1994, 338, 311–318.
- Pepato, M.T.; Migliorini, R.H.; Goldberg, A.L.; Kettelhut, I.C. Role of different proteolytic pathways in degradation of muscle protein from streptozotocin-diabetic rats. Am. J. Physiol. Metab. 1996, 271, E340–E347.
- Price, S.R.; Bailey, J.L.; Wang, X.; Jurkovitz, C.; England, B.K.; Ding, X.; Phillips, L.S.; E Mitch, W. Muscle wasting in insulinopenic rats results from activation of the ATP-dependent, ubiquitin-proteasome proteolytic pathway by a mechanism including gene transcription. J. Clin. Investig. 1996, 98, 1703–1708.
- Tiao, G.; Fagan, J.; Roegner, V.; Lieberman, M.; Wang, J.J.; E Fischer, J.; O Hasselgren, P. Energy-ubiquitin-dependent muscle proteolysis during sepsis in rats is regulated by glucocorticoids. J. Clin. Investig. 1996, 97, 339–348.
- Smith, I.J.; Alamdari, N.; O’Neal, P.; Gonnella, P.; Aversa, Z.; Hasselgren, P.-O. Sepsis increases the expression and activity of the transcription factor Forkhead Box O 1 (FOXO1) in skeletal muscle by a glucocorticoid-dependent mechanism. Int. J. Biochem. Cell Biol. 2010, 42, 701–711.
- Bailey, J.L.; Wang, X.; England, B.K.; Price, S.R.; Ding, X.; Mitch, W.E. The acidosis of chronic renal failure activates muscle proteolysis in rats by augmenting transcription of genes encoding proteins of the ATP-dependent ubiquitin-proteasome pathway. J. Clin. Investig. 1996, 97, 1447–1453.
- Strassburg, S.; Springer, J.; Anker, S.D. Muscle wasting in cardiac cachexia. Int. J. Biochem. Cell Biol. 2005, 37, 1938–1947.
- Gomes, M.D.; Lecker, S.H.; Jagoe, R.T.; Navon, A.; Goldberg, A.L. Atrogin-1, a muscle-specific F-box protein highly expressed during muscle atrophy. Proc. Natl. Acad. Sci. USA 2001, 98, 14440–14445.
- Bodine, S.C.; Latres, E.; Baumhueter, S.; Lai, V.K.; Nunez, L.; Clarke, B.A.; Poueymirou, W.T.; Panaro, F.J.; Na, E.; Dharmarajan, K.; et al. Identification of Ubiquitin Ligases Required for Skeletal Muscle Atrophy. Science 2001, 294, 1704–1708.
- Bodine, S.C.; Baehr, L.M. Skeletal muscle atrophy and the E3 ubiquitin ligases MuRF1 and MAFbx/atrogin-1. Am. J. Physiol. Metab. 2014, 307, E469–E484.
- Cohen, S.; Brault, J.J.; Gygi, S.P.; Glass, D.J.; Valenzuela, D.M.; Gartner, C.; Latres, E.; Goldberg, A.L. During muscle atrophy, thick, but not thin, filament components are degraded by MuRF1-dependent ubiquitylation. J. Cell Biol. 2009, 185, 1083–1095.
- Kedar, V.; McDonough, H.; Arya, R.; Li, H.-H.; Rockman, H.A.; Patterson, C. Muscle-specific RING finger 1 is a bona fide ubiquitin ligase that degrades cardiac troponin I. Proc. Natl. Acad. Sci. USA 2004, 101, 18135–18140.
- Clarke, B.A.; Drujan, D.; Willis, M.S.; Murphy, L.O.; Corpina, R.A.; Burova, E.; Rakhilin, S.V.; Stitt, T.N.; Patterson, C.; Latres, E.; et al. The E3 Ligase MuRF1 Degrades Myosin Heavy Chain Protein in Dexamethasone-Treated Skeletal Muscle. Cell Metab. 2007, 6, 376–385.
- Goll, D.E.; Neti, G.; Mares, S.W.; Thompson, V.F. Myofibrillar protein turnover: The proteasome and the calpains1,2. J. Anim. Sci. 2008, 86, E19–E35.
- Kramerova, I.; Kudryashova, E.; Venkatraman, G.; Spencer, M.J. Calpain 3 participates in sarcomere remodeling by acting upstream of the ubiquitin–proteasome pathway. Hum. Mol. Genet. 2005, 14, 2125–2134.
- Du, J.; Wang, X.; Miereles, C.; Bailey, J.L.; Debigare, R.; Zheng, B.; Price, S.R.; Mitch, W.E. Activation of caspase-3 is an initial step triggering accelerated muscle proteolysis in catabolic conditions. J. Clin. Investig. 2004, 113, 115–123.
- Tidball, J.G.; Spencer, M.J. Expression of a calpastatin transgene slows muscle wasting and obviates changes in myosin isoform expression during murine muscle disuse. J. Physiol. 2002, 545, 819–828.
- Huang, J.; Forsberg, N.E. Role of calpain in skeletal-muscle protein degradation. Proc. Natl. Acad. Sci. USA 1998, 95, 12100–12105.
- Purintrapiban, J.; Wang, M.-C.; Forsberg, N.E. Degradation of sarcomeric and cytoskeletal proteins in cultured skeletal muscle cells. Comp. Biochem. Physiol. Part B Biochem. Mol. Biol. 2003, 136, 393–401.
- Goldbraikh, D.; Neufeld, D.; Eid-Mutlak, Y.; Lasry, I.; E Gilda, J.; Parnis, A.; Cohen, S. USP 1 deubiquitinates Akt to inhibit PI 3K-Akt-FoxO signaling in muscle during prolonged starvation. EMBO Rep. 2020, 21, e48791.
- Coyne, E.S.; Bedard, N.; Wykes, L.; Stretch, C.; Jammoul, S.; Li, S.; Zhang, K.; Sladek, R.S.; Bathe, O.F.; Jagoe, R.T.; et al. Knockout of USP19 Deubiquitinating Enzyme Prevents Muscle Wasting by Modulating Insulin and Glucocorticoid Signaling. Endocrinology 2018, 159, 2966–2977.
- Cohen, S.; Lee, D.; Zhai, B.; Gygi, S.P.; Goldberg, A.L. Trim32 reduces PI3K-Akt-FoxO signaling in muscle atrophy by promoting plakoglobin-PI3K dissociation. J. Cell Biol. 2014, 204, 747–758.
- Chen, L.; Huang, J.; Ji, Y.; Zhang, X.; Wang, P.; Deng, K.; Jiang, X.; Ma, G.; Li, H.-L. Tripartite motif 32 prevents pathological cardiac hypertrophy. Clin. Sci. 2016, 130, 813–828.
- Sandri, M. Signaling in Muscle Atrophy and Hypertrophy. Physiology 2008, 23, 160–170.
- Agnetti, G.; Halperin, V.L.; Kirk, J.A.; Chakir, K.; Guo, Y.; Lund, L.; Nicolini, F.; Gherli, T.; Guarnieri, C.; Caldarera, C.M.; et al. Desmin modifications associate with amyloid-like oligomers deposition in heart failure. Cardiovasc. Res. 2014, 102, 24–34.
- Shieh, S.-Y.; Ikeda, M.; Taya, Y.; Prives, C. DNA Damage-Induced Phosphorylation of p53 Alleviates Inhibition by MDM2. Cell 1997, 91, 325–334.
- Lin, D.I.; Barbash, O.; Kumar, K.S.; Weber, J.D.; Harper, J.W.; Klein-Szanto, A.J.P.; Rustgi, A.; Fuchs, S.Y.; Diehl, J.A. Phosphorylation-Dependent Ubiquitination of Cyclin D1 by the SCFFBX4-αB Crystallin Complex. Mol. Cell 2006, 24, 355–366.
- Koepp, D.M.; Schaefer, L.K.; Ye, X.; Keyomarsi, K.; Chu, C.; Harper, J.W.; Elledge, S.J. Phosphorylation-dependent ubiquitination of cyclin E by the SCFFbw7 ubiquitin ligase. Science 2001, 294, 173–177.
- Hart, M.; Concordet, J.-P.; Lassot, I.; Albert, I.; Santos, R.D.L.; Durand, H.; Perret, C.; Rubinfeld, B.; Margottin, F.; Benarous, R.; et al. The F-box protein β-TrCP associates with phosphorylated β-catenin and regulates its activity in the cell. Curr. Biol. 1999, 9, 207–211.
- Shivanna, S.; Harrold, I.; Shashar, M.; Meyer, R.; Kiang, C.; Francis, J.; Zhao, Q.; Feng, H.; Edelman, E.R.; Rahimi, N.; et al. The c-Cbl Ubiquitin Ligase Regulates Nuclear β-Catenin and Angiogenesis by Its Tyrosine Phosphorylation Mediated through the Wnt Signaling Pathway. J. Biol. Chem. 2015, 290, 12537–12546.
- Nakagawa, T.; Yokoe, S.; Asahi, M. Phospholamban degradation is induced by phosphorylation-mediated ubiquitination and inhibited by interaction with cardiac type Sarco(endo)plasmic reticulum Ca2+-ATPase. Biochem. Biophys. Res. Commun. 2016, 472, 523–530.
- Geisler, N.; Weber, K. The amino acid sequence of chicken muscle desmin provides a common structural model for intermediate filament proteins. EMBO J. 1982, 1, 1649–1656.
- Milner, D.J.; Weitzer, G.; Tran, D.; Bradley, A.; Capetanaki, Y. Disruption of muscle architecture and myocardial degeneration in mice lacking desmin. J. Cell Biol. 1996, 134, 1255–1270.
- Diokmetzidou, A.; Soumaka, E.; Kloukina, I.; Tsikitis, M.; Makridakis, M.; Varela, A.; Davos, C.H.; Georgopoulos, S.; Anesti, V.; Vlahou, A.; et al. Desmin and αB-crystallin interplay in the maintenance of mitochondrial homeostasis and cardiomyocyte survival. J. Cell Sci. 2016, 129, 3705–3720.
- Goldfarb, L.G.; Olivé, M.; Vicart, P.; Goebel, H.H. Intermediate Filament Diseases: Desminopathy. Cannabinoids Neuropsychiatr. Disord. 2008, 642, 131–164.
- Ma, X.-W.; Li, Q.; Xu, P.-T.; Zhang, L.; Li, H.; Yu, Z.-B. Tetanic contractions impair sarcomeric Z-disk of atrophic soleus muscle via calpain pathway. Mol. Cell. Biochem. 2011, 354, 171–180.
- Benson, D.W.; O Hasselgren, P.; Hiyama, D.T.; James, J.H.; Li, S.; Rigel, D.F.; E Fischer, J. Effect of sepsis on calcium uptake and content in skeletal muscle and regulation in vitro by calcium of total and myofibrillar protein breakdown in control and septic muscle: Results from a preliminary study. Surgery 1989, 106, 87–93.
- Fischer, D.R.; Sun, X.; Williams, A.B.; Gang, G.; Pritts, T.A.; James, H.J.; Molloy, M.; Fischer, J.E.; Paul, R.J.; Hasselgren, P.-O. Dantrolene reduces serum tnfα and corticosterone levels and muscle calcium, calpain gene expression, and protein breakdown in septic rats. Shock 2001, 15, 200–207.
- Costelli, P.; Bossola, M.; Muscaritoli, M.; Grieco, G.; Bonelli, G.; Bellantone, R.; Doglietto, G.; Baccino, F.; Fanelli, F.R. Anticytokine treatment prevents the increase in the activity of atp-ubiquitin- and ca2+-dependent proteolytic systems in the muscle of tumour-bearing rats. Cytokine 2002, 19, 1–5.
- Turner, P.; Schultz, R.; Ganguly, B.; Steinhardt, R. Proteolysis results in altered leak channel kinetics and elevated free calcium in mdx muscle. J. Membr. Biol. 1993, 133, 243–251.
- Whitehead, N.P.; Yeung, E.W.; Allen, D.G. Muscle damage in mdx (dystrophic) mice: Role of calcium and reactive oxygen species. Clin. Exp. Pharmacol. Physiol. 2006, 33, 657–662.
- Ye, X.; Zhang, H.M.; Qiu, Y.; Hanson, P.J.; Hemida, M.G.; Wei, W.; Hoodless, P.A.; Chu, F.; Yang, D. Coxsackievirus-Induced miR-21 Disrupts Cardiomyocyte Interactions via the Downregulation of Intercalated Disk Components. PLoS Pathog. 2014, 10, e1004070.
- Sagar, G.D.V.; Gereben, B.; Callebaut, I.; Mornon, J.-P.; Zeöld, A.; Da Silva, W.S.; Luongo, C.; Dentice, M.; Tente, S.M.; Freitas, B.C.G.; et al. Ubiquitination-Induced Conformational Change within the Deiodinase Dimer Is a Switch Regulating Enzyme Activity. Mol. Cell. Biol. 2007, 27, 4774–4783.
- Watkins, G.R.; Wang, N.; Mazalouskas, M.D.; Gomez, R.J.; Guthrie, C.R.; Kraemer, B.C.; Schweiger, S.; Spiller, B.W.; Wadzinski, B.E. Monoubiquitination Promotes Calpain Cleavage of the Protein Phosphatase 2A (PP2A) Regulatory Subunit α4, Altering PP2A Stability and Microtubule-associated Protein Phosphorylation. J. Biol. Chem. 2012, 287, 24207–24215.
- Twomey, C.; Qian, S.; McCarthy, J.V. TRAF6 promotes ubiquitination and regulated intramembrane proteolysis of IL-1R1. Biochem. Biophys. Res. Commun. 2009, 381, 418–423.
- Bar-Nun, S.; Glickman, M.H. Proteasomal AAA-ATPases: Structure and function. Biochim. Biophys. Acta (BBA) Bioenerg. 2012, 1823, 67–82.
- Piccirillo, R.; Goldberg, A.L. The p97/VCP ATPase is critical in muscle atrophy and the accelerated degradation of muscle proteins. EMBO J. 2012, 31, 3334–3350.