The cystine/glutamate antiporter xCT is a tumor-associated antigen that has been newly identified in many cancer types. By participating in glutathione biosynthesis, xCT protects cancer cells from oxidative stress conditions and ferroptosis, and contributes to metabolic reprogramming, thus promoting tumor progression and chemoresistance. Moreover, xCT is overexpressed in cancer stem cells. These features render xCT a promising target for cancer therapy, as has been widely reported in the literature and in our work on its immunotargeting. Interestingly, studies on the TP53 gene have revealed that both wild-type and mutant p53 induce the post-transcriptional down- modulation of xCT, contributing to ferroptosis. Moreover, APR-246, a small molecule drug that can restore wild-type p53 function in cancer cells, has been described as an indirect modulator of xCT expression in tumors with mutant p53 accumulation, and is thus a promising drug to use in combination with xCT inhibition.
- Tumor-Associated Antigen xCT
- p53
- APR-246
- PRIMA-1
- Cancer immunotherpy
1. Introduction
xCT overexpression in many cancer histotypes suggests that it may have other key roles in cancer survival and progression beyond its well documented function in maintaining redox balance and in counteracting oxidative damage through GSH synthesis.
Studies in a number of glioma cell lines have reported that the expression of xCT and the regulatory subunit 4F2hc causes glutamate release, which can activate glutamate receptors, such as the ionotropic α-amino-3-hydroxyl-5-methyl-4-isoxazole-propionate (AMPA) receptor, in an autocrine or paracrine manner, causing glioma cell invasion and peritumoral excitotoxic neuronal cell loss [1]. More recently, Dornier et al. [2] have shown that both invasive MMTV-PyMT breast tumor-derived cells and MDA-MB-231 human cells release more glutamate than normal breast epithelial cells thanks to the acquisition of xCT expression. Furthermore, it has been demonstrated that the invasive protrusions observed in these cells depend on the presence of glutamine but not glucose in the culture medium, whereas glutamine deprivation reduces protrusion length. This indicates that glutaminolysis is responsible for invasiveness. Glutamate release activates the metabotropic glutamate receptor (GRM) that is expressed on cancer cells, which promotes Rab27 GTPase activity, which, in turn, mediates the trafficking of MT1-MMP metalloprotease on the cell membrane. The pharmacological inhibition of xCT with sulfasalazine (SAS) in vitro [1][2] and in vivo [1], via siRNA knockdown [2], or via the inhibition of the glutamate receptors or their downstream effectors [1][2], reduces the migration and invasiveness of cancer cells both in breast and glioma cancer cells. Further evidence for the involvement of xCT in metastasis formation comes from studies on xCTKO melanocytes, in which the caveolin 1/β-catenin pathway, which is responsible for cell–cell adhesion, is activated as a consequence of p38-mediated caveolin 1 upregulation after ROS accumulation. This promotes the translocation of β-catenin from the nucleus to the cell membrane [3], thus inhibiting metastasis. On the other hand, xCT overexpression in glioblastoma cells confers resistance to oxidative stress induction and favors anchorage-independent cell growth, rendering them more tumorigenic [4].
In light of its function as an amino acid transporter, it is not surprising that xCT is implicated in the metabolic reprogramming of cancer cells and adaptation to nutrient availability in the tumor microenvironment, and many studies have uncovered an unexpected weakness in cancer cells cultured under glucose deprivation conditions [5][6][7]. Indeed, this may appear logical if we suppose that the effects of glucose starvation can be prevented by xCT’s detoxification of induced ROS. However, it has been demonstrated in several cell lines that xCT activity is responsible for glucose starvation-induced cell death [5][6][7]. In fact, xCT expression causes cell death upon glucose withdrawal, rendering cancer cells glucose-addicted. These effects are rescued when xCT is downregulated or pharmacologically inhibited with SAS. xCT amino acid transport, and not its antioxidant function, seems to be responsible for cell death upon glucose starvation, since xCT upregulation occurs independently of ROS generation under these conditions [6]. However, it is still unclear whether cystine uptake or glutamate export is the main cause of glucose addiction in cancer cells with elevated xCT expression. Indeed, both glucose and glutamine are considered to be important nutrients for cancer cell growth [6][7]. When glucose is depleted, cancer cells become more dependent on glutamine, which is converted to glutamate to produce α-ketoglutarate (α-KG), a key intermediate in the tricarboxylic acid (TCA) cycle, which generates substrates for oxidative phosphorylation (OXPHOS) to produce energy. If an excess of glutamate is exported due to xCT overexpression, the levels of α-KG drop, and a concomitant shortage of glucose leads to the collapse of the TCA cycle and mitochondrial respiration. Moreover, the increased xCT activity triggers increased nicotinamide adenine dinucleotide phosphate (NADPH) consumption in the reduction of cystine to cysteine. Upon glucose deprivation, the pentose phosphate pathway is blunted, and NADPH cannot be efficiently regenerated. This unbalance between NADPH consumption and regeneration ultimately leads to an impairment in the capacity to buffer cellular ROS [8]. This has been confirmed in all of the cited studies, as the addition of α-KG greatly improved cell viability under glucose starvation conditions [5][6][7]. interestingly, xCT expression during glucose withdrawal is dependent on NRF2 and ATF4 activation [6][7].
Cell metabolism reprogramming can make xCT-expressing cancer cells dependent on glutamine, as demonstrated in Non-Small Cell Lung Cancer (NSCLC) cells [9]. Indeed, xCT overexpression leads to higher consumption of cystine and other amino acids, and significant glutamate release. xCT-overexpressing lung cancer cells are more sensitive to glutamine withdrawal compared to xCT-knockdown cells and show a less invasive phenotype in the absence of glutamine, indicating that glutaminolysis becomes an essential metabolic pathway in the presence of high xCT levels. In general, these cells show higher glucose consumption as well as higher glutamine and lactate production, with the upregulation of OXPHOS, all hallmarks of metabolic cancer cell reprogramming [10].
Finally, the altered metabolic state caused by xCT expression also has consequences on mitochondrial functions, with mitochondrial genes being upregulated because of the increased OXPHOS [4]. Overall, these studies provide evidence that supports the key role played by xCT in both redox homeostasis and nutrient dependency in cancer cells, as discussed in detail by Koppula et al. [11].
2. xCT Targeting and APR-246 Administration: A New Therapeutic Combinatorial Strategy
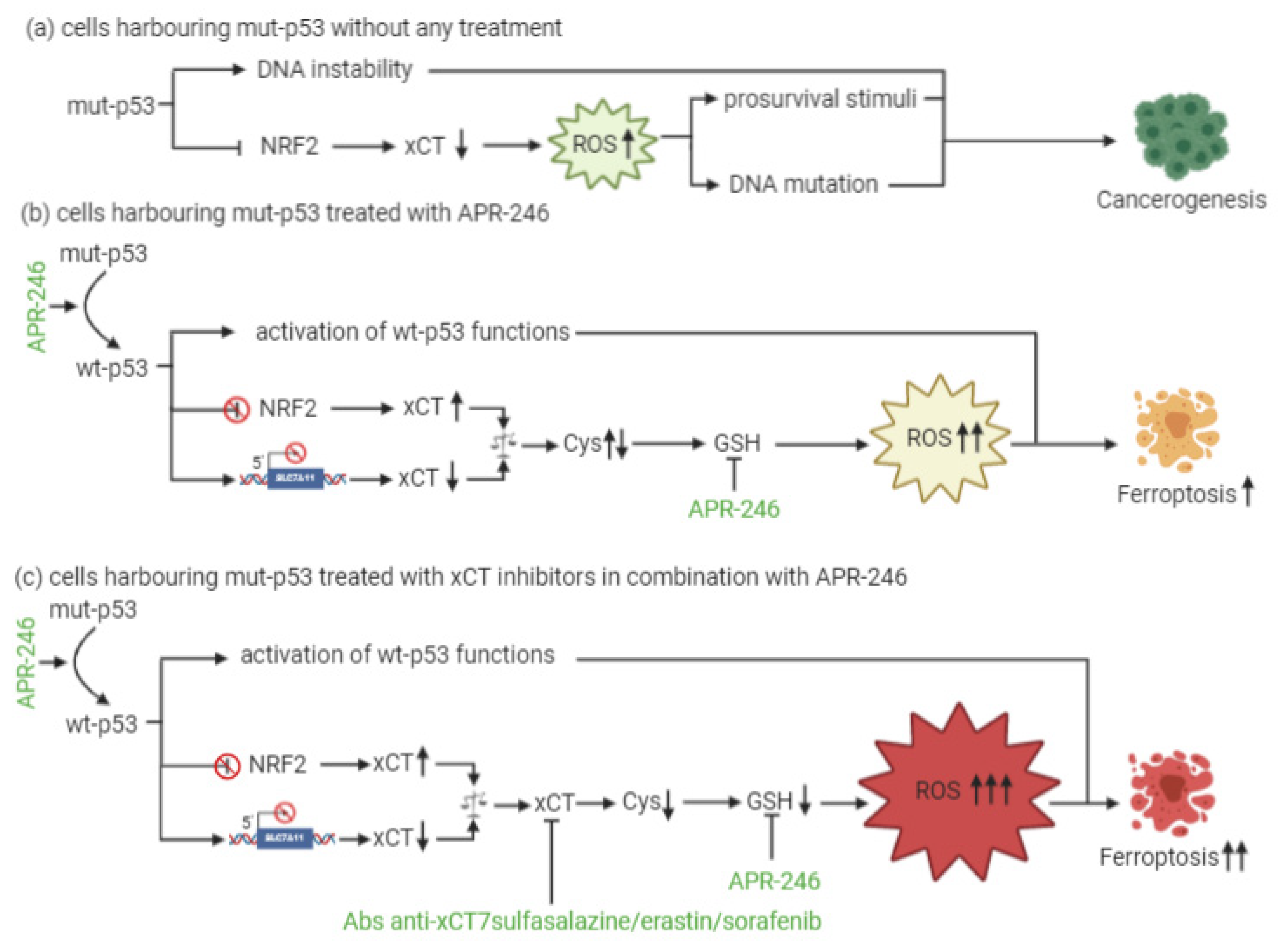
At present, little is known about a combinatorial approach that includes xCT targeting and p53 reactivation through APR-246 treatment. The dual mechanism of action of APR-246, which includes a role in GSH and ROS modulation together with its effects on mut-p53, is, however, quite well established. In particular, the active component of APR-246 has been shown to react with GSH, forming an adduct (GSH-MQ) that depletes GSH from the cells, stopping its recycling by GSH reductase [12]. This leads to ROS accumulation, especially in mitochondria, causing lipid peroxidation, mitochondrial rupture, and cytochrome-c release. In mut-p53 osteosarcoma cells, APR-246 can inhibit thioredoxin-1 (Trx1) and glutaredoxin-1 (Grx1), which are commonly upregulated in cancer cells, and favor their protection against oxidative stress [13]. Moreover, the treatment of breast cancer cells with APR-246 for 12 h causes perturbations in gene expression, with six genes involved in ROS modulation being differentially expressed in all of the cell lines investigated. SLC7A11 is among these genes [14]. Therefore, these results have revealed the dual role that APR-246 plays as a reactivator of mut-p53 and an inducer of ROS accumulation in cancer cells. This latter mechanism might explain why this drug can even exert anticancer activity in wt-p53 cells.
In the context of p53-mediated modulation over xCT expression and activity, Liu and colleagues have shown for the first time that xCT blockade and APR-246 administration may act synergistically to target and kill mut-p53 esophageal cancer cells [12]. As a result of APR-246 treatment, ROS levels increase and GSH cellular stores become eroded, and the attenuated xCT expression, resulting from mut-p53-mediated NRF2 inhibition, further reduces the ability of cancer cells to cope with oxidative stress. However, similarly to what was described by Jiang and colleagues in the context of wt-p53 [15], the actual levels of xCT expression are crucial for the sensitivity of mut-p53 cells to oxidative damage, and the expression level of xCT may serve as a predictive biomarker of the response of mut-p53 cancers to APR-246. In particular, cells with low xCT levels have proven to be the most sensitive to the treatment.
Remarkably, the therapeutic efficacy of APR-246 in mut-p53 harboring cancer cells can be sharply increased by concomitant xCT inhibition, leading to the synergistic induction of ROS accumulation and apoptosis [12]. The rationale for such a combinatorial strategy is further supported by the notion that APR-246 disrupts the interaction between mut-p53 and NRF2 [16]. Therefore, it has been proposed that this can release NRF2 from mut-p53 inhibition restoring elevated xCT levels, and potentially antagonizing the therapeutic activity of APR-246, thanks to GSH depletion [17]. Based on these findings, the increased therapeutic efficacy of the combination of APR-246 and xCT inhibitors is unlikely to be the result of the restoration of wt-p53 functions with respect to SLC7A11 gene expression, since wt-p53 should also lead to its transcriptional repression, albeit through a distinct molecular mechanism, but rather it appears to depend on APR-246 oxidative stress induction (Figure 1). Overall, these lines of evidence suggest that the predominant mechanism underlying APR-246 therapeutic activity relies on thean impairment of cancer cell antioxidant defenses, an effect that can be dramatically augmented in the context of xCT inhibition.
References
- Lyons, S.A.; Chung, W.J.; Weaver, A.K.; Ogunrinu, T.; Sontheimer, H. Autocrine glutamate signaling promotes glioma cell invasion. Cancer Res. 2007, 67, 9463–9471.
- Dornier, E.; Rabas, N.; Mitchell, L.; Novo, D.; Dhayade, S.; Marco, S.; MacKay, G.; Sumpton, D.; Pallares, M.; Nixon, C.; et al. Glutaminolysis drives membrane trafficking to promote invasiveness of breast cancer cells. Nat. Commun. 2017, 8, 1–14.
- Chen, R.; Song, Y.; Zhou, Z.; Tong, T.; Li, Y.; Fu, M.; Guo, X.; Dong, L.; He, X.; Qiao, H.; et al. Disruption of xCT inhibits cancer cell metastasis via the caveolin-1 /beta-catenin pathway. Oncogene 2009, 28, 599–609.
- Polewski, M.D.; Reveron, T.R.F.; Cherryholmes, G.A.; Marinov, G.K.; Cassady, K.; Aboody, K.S. Increased expression of system xc- in glioblastoma confers an altered metabolic state and temozolomide resistance. Mol. Cancer Res. 2016, 14, 1229–1242.
- Goji, T.; Takahara, K.; Negishi, M.; Katoh, H. Cystine uptake through the cystine/glutamate antiporter xCT triggers glioblastoma cell death under glucose deprivation. J. Biol. Chem. 2017, 292, 19721–19732.
- Koppula, P.; Zhang, Y.; Shi, J.; Li, W.; Gan, B. The glutamate/cystine antiporter SLC7A11/xCT enhances cancer cell dependency on glucose by exporting glutamate. J. Biol. Chem. 2017, 292, 14240–14249.
- Shin, C.S.; Mishra, P.; Watrous, J.D.; Carelli, V.; Aurelio, M.; Jain, M.; Chan, D.C. The glutamate/cystine xCT antiporter antagonizes glutamine metabolism and reduces nutrient flexibility. Nat. Commun. 2017, 8, 1–11.
- Liu, X.; Olszewski, K.; Zhang, Y.; Lim, E.W.; Shi, J.; Zhang, X.; Zhang, J.; Lee, H.; Koppula, P.; Lei, G.; et al. Cystine transporter regulation of pentose phosphate pathway dependency and disulfide stress exposes a targetable metabolic vulnerability in cancer. Nat. Cell Biol. 2020, 22, 476–486.
- Ji, X.; Qian, J.; Rahman, S.M.J.; Siska, P.J.; Zou, Y.; Harris, B.K.; Hoeksema, M.D.; Trenary, I.A.; Heidi, C.; Eisenberg, R.; et al. xCT (SLC7A11)-mediated metabolic reprogramming promotes non-small cell lung cancer progression. Oncogene 2018, 37, 5007–5019.
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674.
- Koppula, P.; Zhang, Y.; Zhuang, L.; Gan, B. Amino acid transporter SLC7A11/xCT at the crossroads of regulating redox homeostasis and nutrient dependency of cancer. Cancer Commun. (Lond. Engl.) 2018, 38, 12.
- Liu, D.S.; Duong, C.P.; Haupt, S.; Montgomery, K.G.; House, C.M.; Azar, W.J.; Pearson, H.B.; Fisher, O.M.; Read, M.; Guerra, G.R.; et al. Inhibiting the system xC-/glutathione axis selectively targets cancers with mutant-p53 accumulation. Nat. Commun. 2017, 8, 1–14.
- Haffo, L.; Lu, J.; Bykov, V.J.N.; Martin, S.S.; Ren, X.; Coppo, L.; Wiman, K.G.; Holmgren, A. Inhibition of the glutaredoxin and thioredoxin systems and ribonucleotide reductase by mutant p53-targeting compound APR-246. Sci. Rep. 2018, 8, 1–10.
- Naoise, C.; Synnott, S.F.M.; Vladimir, J.N.; Bykov, J.; Crown, K.G.W.; Michael, J.D. The Mutant p53-Targeting Compound APR-246 Induces ROS-Modulating Genes in Breast Cancer Cells. Transl. Oncol. 2018, 11, 1343–1349.
- Jiang, L.; Kon, N.; Li, T.; Wang, S.J.; Su, T.; Hibshoosh, H.; Baer, R.; Gu, W. Ferroptosis as a p53-mediated activity during tumour suppression. Nature 2015, 520, 57–62.
- Walerych, D.; Lisek, K.; Sommaggio, R.; Piazza, S.; Ciani, Y.; Dalla, E.; Rajkowska, K.; Gaweda, W.K.; Ingallina, E.; Tonelli, C.; et al. Proteasome machinery is instrumental in a common gain-of-function program of the p53 missense mutants in cancer. Nat. Cell Biol. 2016, 18, 897–909.
- Clemons, N.J.; Liu, D.S.; Duong, C.P.; Phillips, W.A. Inhibiting system xC− and glutathione biosynthesis–a potential Achilles’ heel in mutant-p53 cancers. Mol. Cell. Oncol. 2017, 4.