Lipid rafts are dynamic and shifting assemblies of sphingolipids, cholesterol, glycosphingolipids, and proteins forming platforms or lipid microdomains for the organization and dynamic contact of molecules involved in several molecular and cellular processes such as ligand affinity, effector specificity, signal transduction, membrane sorting, and receptor trafficking and recycling.
- lipid rafts
- dopamine receptor
1. Structure of Lipid Rafts
Cholesterol molecules rest between the hydrophobic tails of phospholipids; next to them, many proteins serve as anchor units for other proteins. The organization of certain groups of proteins allows optimal function, as well as specification of cellular signal transmission by granting effective interaction between proteins and avoiding interference of opposite signaling pathways [2][6][2,6]. Lipid rafts are located in cell surface membranes, as well as in intracellular membranes. These allow the coordination of cellular processes, which in turn affect bioactivity of many receptors and the cell itself [2].
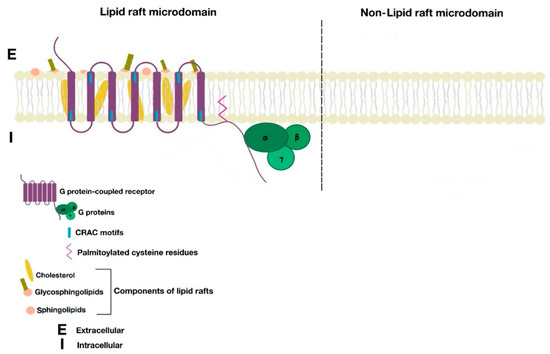
Figure 1. Cell membrane partitions into lipid raft and non-lipid raft microdomains.
Many proteins are associated with lipid rafts; one of the best characterized proteins are the caveolins. These form caveolae (“little caves”). Caveolae (Figure 2) are scaffolding proteins that assemble within lipid raft domains to form flask-shaped invaginations of the plasma membrane. These platforms allow protein–protein, lipid–lipid, and protein–lipid interactions and regulate numerous cellular processes (signal transduction, receptor trafficking, recycling, etc.). There are three caveolin isoforms (Cav1, Cav2, and Cav3). However, Cav1 is the isoform that is present in most tissues and is enough for the formation of caveolae [7].
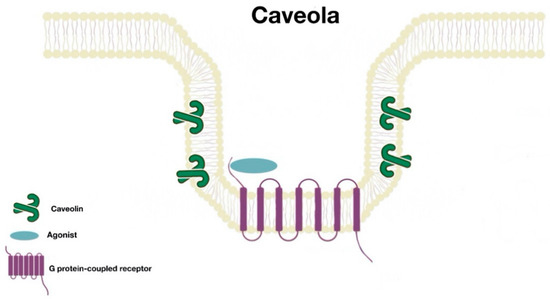
Figure 2. Caveola structure.
2. Function
The function of surface membrane receptors is determined by their intracellular trafficking pathway. These receptors are produced intracellularly and their trafficking to the plasma membrane is needed for the attachment to the extracellular agonist or antagonist. Agonist-induced activation of the receptor is followed by its subsequent internalization and re-insertion to the plasma membrane (desensitization and resensitization of the receptor), ultimately controlling the function and activity of the receptor [7][8][9][10][11][7,8,9,10,11].
3. Associated Components
G protein-coupled receptors (GPCRs) are the largest superfamily of mammalian surface membrane receptors that work as signaling proteins. These are involved in diverse physiological processes by binding to ligands and transducing intracellular signaling pathways via activation of G proteins [9][10][11][9,10,11], i.e., G protein subunits α, β, and γ. The structure of GPCRs features the presence of seven transmembrane α-helical molecules, which are connected by three extracellular and three intracellular loops [10].
Surface membrane protein receptors, including GPCRs, undergo a trafficking process with constant co-translational and post-translational changes before arriving at the plasma membrane. As with many cellular proteins, GPCRs, following their synthesis, reside in the endoplasmic reticulum (ER) where they undergo assembly, folding, and packaging. The GPCRs temporarily reside in endoplasmic reticulum-derived COPII (coat protein complex II) transport vesicles, before exiting the ER into the Golgi apparatus. There, the GPCRs undergo further modifications, such as the S-palmitoylation of cysteine residues. At the Golgi edge, the GPCRs are boxed again into vesicles, into the endosomal pathway, which targets the receptors to the plasma membrane. In the plasma membrane, the GPCRs mature and become functional. Some subsets of GPCRs are targeted to specific regions of the plasma membrane, such as the lipid rafts [11].
There are many motifs of proteins in the GPCRs that interact with the plasma membrane and its associated cholesterol. These motifs carry a cholesterol recognition/interaction amino acid consensus or CRAC motif. The CRAC motif helps anchor the Cav isoforms to the plasma membrane [12] [12] as well as other plasma proteins, including GPCRs. Once the GPCR is targeted to a lipid raft, it can be successfully stimulated by interacting with its ligand (Figure 3). The agonist occupation of GPCRs, as in dopamine receptors, results in a conformational change promoting the exchange of GDP (guanosine diphosphate) to GTP (guanosine triphosphate) on the Gα subunit of the G protein, leading to two actions (Figure 4). First, the uncoupling of the G proteins from the GPCR, and second, the uncoupling of the Gα subunit from the Gβγ subunit. Finally, the Gα subunit stimulates or inactivates downstream signaling processes, depending on its conformational and functional characterization, Gαs being stimulatory or Gαi being inhibitory of adenylyl cyclase activity. The remaining Gβγ subunit activates GPCR kinases (GRKs), which phosphorylate amino acids in the third intracellular loop of the transmembrane segments and C-terminal tail of the receptor, leading to the activation of cytosolic proteins known as arrestins [13][14] [13,14] (Figure 5). The activation of GRKs and β-arrestins ensures the removal of the GPCR from the cell surface, leading to its desensitization.
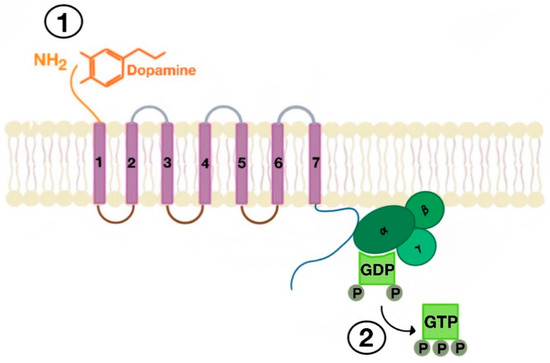
Figure 3. Agonist-induced stimulation of G protein-coupled receptors (GPCRs) (e.g., dopamine receptor (1) and phosphorylation of GDP to GTP on the Gα subunit of the G protein (2)).
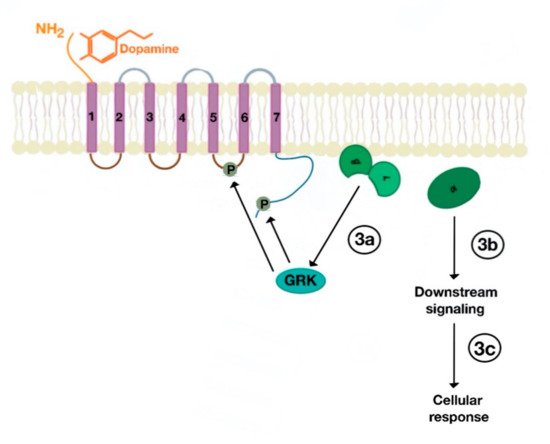
Figure 4. Uncoupling of Gα subunit from Gβγ subunit, followed by activation GPCR kinases (GRKs), which phosphorylate amino acids in the third intracellular loop of the transmembrane segments and C-terminal tail (3a). The uncoupled Gα subunit activates downstream cellular (3b) and subsequent cellular response (3c).
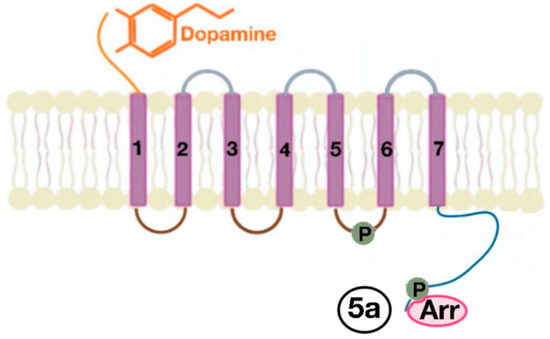
Figure 5. The phosphorylation of amino acids in the third intracellular loop of the transmembrane segments and C-terminal tail leads to the activation of β-arrestins (5a).
The β-arrestins, originally discovered for their inhibitory part in some receptor signaling pathways, have been proven to work as critical controllers of agonist-induced endocytosis and ubiquitination of plasma receptors, including GPCRs. Therefore, β-arrestins play a pivotal role in the trafficking path of endocytosed GPCRs [13][14][13,14].
The β-arrestins direct sequestration of GPCRs via a dynamin-dependent, clathrin-mediated endocytosis. This process takes effect through adaptor protein 2 (AP2) complexes, which in turn begin clathrin-coated pit assembly (Figure 6). As soon as the GPCR is internalized, it is packaged in early endosome (Figure 7), where the GPCR is dephosphorylated by protein phosphatase 2A, in the case of the D1R [14]. Then, sorting nexins (SNXs) distribute the GPCR to different destinations [15]. From the endosome, the receptor is either sent to fast recycling endosome and subsequently to the lipid raft in the plasma membrane, or to lysosomes (Figure 8) for eventual degradation.
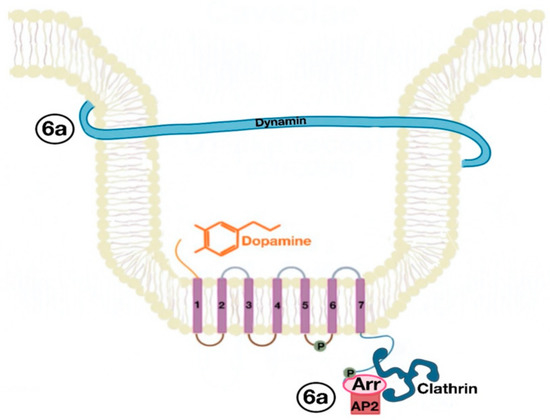
Figure 6. Dynamin and clathrin associate with the GPCR (dopamine receptor) and mediate its endocytosis by means of the adaptor protein 2 (AP2) (6a).
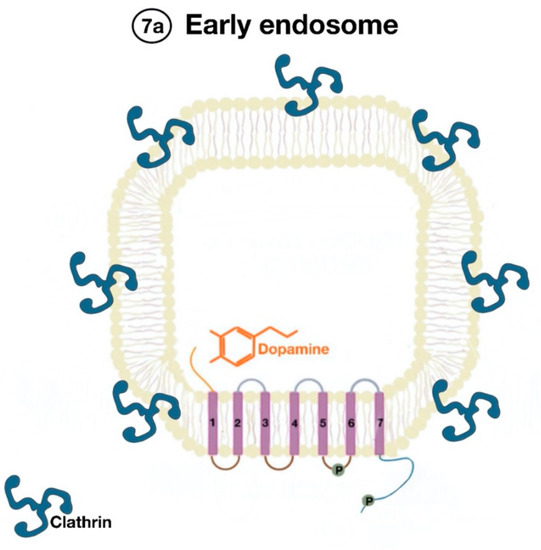
Figure 7. Endocytosed GPCR (dopamine receptor) is packaged in an early endosome (7a).
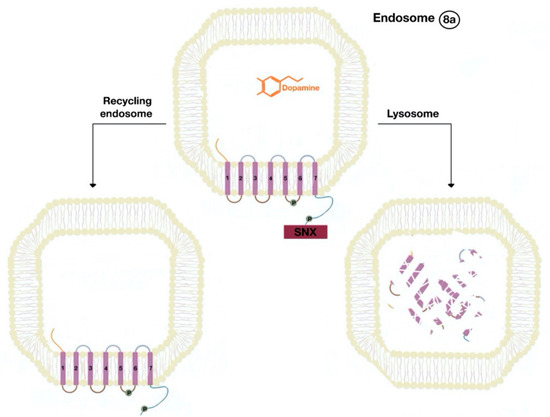
Figure 8. The endocytosed GPCR (dopamine receptor) binds to sorting nexin (SNX) that sends the GPCR either to a recycling endosome or lysosome (8a). Receptors targeted to lysosomes are degraded.
The SNXs are a diverse group of membrane-linked phosphoinositide-binding proteins that regulate cellular protein trafficking by controlling all aspects of the endocytic pathway, including endocytosis, endosomal sorting, and endosomal signaling [16]. The SNX either sorts the endocytosed and dephosphorylated/resensitized GPCR into the fast recycling endosomes, targeting it to lipid rafts (Figure 9), or send such receptor to late endosomes, targeting it to lysosomes, inducing the inevitable degradation of the GPCR [17].
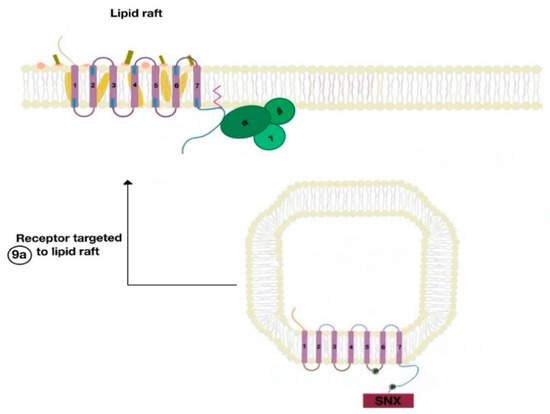
Figure 9. The resensitized GPCR (dopamine receptor) is targeted to the lipid raft microdomain by SNX (9a) and is ready to bind to a new agonist (9a).