In 2015 the European Respiratory Society (ERS) and the American Thoracic Society (ATS) “Task Force on Undifferentiated Forms of Connective Tissue Disease-associated Interstitial Lung Disease” proposed classification criteria for a new research category defined as “Interstitial Pneumonia with Autoimmune Features” (IPAF), to uniformly define patients with interstitial lung disease (ILD) and features of autoimmunity, without a definite connective tissue disease. These classification criteria were based on a variable combination of features obtained from three domains: a clinical domain consisting of extra-thoracic features, a serologic domain with specific autoantibodies, and a morphologic domain with imaging patterns, histopathological findings, or multicompartment involvement. Features suggesting a systemic vasculitis were excluded. Since publication of ERS/ATS IPAF research criteria, various retrospective studies have been published focusing on prevalence; clinical, morphological, and serological features; and prognosis of these patients showing a broad heterogeneity in the results. Recently, two prospective, cohort studies were performed, confirming the existence of some peculiarities for this clinical entity and the possible progression of IPAF to a defined connective tissue disease (CTD) in about 15% of cases. Moreover, a non-specific interstitial pneumonia pattern, an anti-nuclear antibody positivity, and a Raynaud phenomenon were the most common findings. In comparison with idiopathic pulmonary fibrosis (IPF), IPAF patients showed a better performance in pulmonary function tests and less necessity of oxygen delivery. However, at this stage of our knowledge, we believe that further prospective studies, possibly derived from multicenter cohorts and through randomized control trials, to further validate the proposed classification criteria are needed.
- Interstitial Pneumonia with Autoimmune Features
1. Introduction
Interstitial lung diseases (ILDs) refer to a broad category of more than 200 lung diseases including a variety of illnesses with diverse causes, treatments, and prognoses. These disorders are grouped together because of similarities in their clinical presentation, plain chest radiographic appearance, and physiologic features leading ultimately—at least in a number of cases—to pulmonary fibrosis. ILDs contain several categories, characterized by different prognoses—including idiopathic interstitial pneumonias (IIPs) and connective tissue disease (CTD)-associated interstitial lung disease (CTD-ILD)[1].
The IIPs are a group of heterogeneous disorders characterized by diffuse parenchymal lung involvement with overlapping clinical and radiologic features[2]. They are generally categorized by histopathologic pattern, and the term chronic fibrosing interstitial pneumonia (IP) has recently been used to encompass the histopathologic patterns of usual interstitial pneumonia (UIP) and nonspecific interstitial pneumonia (NSIP)[1].
However, in clinical practice, it is common to come across patients with an “idiopathic” interstitial pneumonia (IIP) associated with features suggestive of, but not diagnostic for, a classical CTD [5][6].
On the basis of previous studies, in 2015 the European Respiratory Society (ERS) and the American Thoracic Society (ATS) “Task Force on Undifferentiated Forms of Connective Tissue Disease-associated Interstitial Lung Disease” proposed classification criteria for a new research category defined as “Interstitial Pneumonia with Autoimmune Features” (IPAF). The classification of IPAF can therefore be considered an overlap between an idiopathic interstitial pneumonia and CTD-ILDs[7].
2. Criteria for Interstitial Pneumonia with Autoimmune Features: The European Respiratory Society/American Thoracic Society Research Statement
As discussed above, there is agreement that some patients with an idiopathic ILD may have some features that suggest the presence of a systemic autoimmune process but do not meet classification criteria for a defined CTD[8][9][10]. Therefore, it is common to have discordance among specialists about how to diagnose such patients. A correct identification of patients with CTD-ILD can be challenging if the lung is the predominant or the primary organ involved and clinical evidence of a systemic autoimmune disease is subtle or absent[9].
Moreover, different IIPs may have different prognoses and the available therapeutic options may significantly differ[11][12][13].
In contrast to the adverse effects observed in patients with IPF [14], CTD-ILD could benefit from immunosuppressive treatment, including steroids[15][16]. On the other hand, patients with IPF benefit from antifibrotic agents[17][18], currently under investigation in CTD-ILD patients[19][20]. The lack of consensus over nomenclature and classification criteria limits the ability to perform prospective studies in patients with this particular subset of interstitial pneumonia.
Between 2010 and 2012, different studies proposed different, but partially overlapping, criteria and terms to describe these patients, including “undifferentiated CTD-associated ILD” (UCTD-ILD) [21][22], “lung-dominant CTD” [23] or “autoimmune-featured ILD”[24]. However, each term was capturing different patients and, therefore, none has been universally accepted.
Table 1.
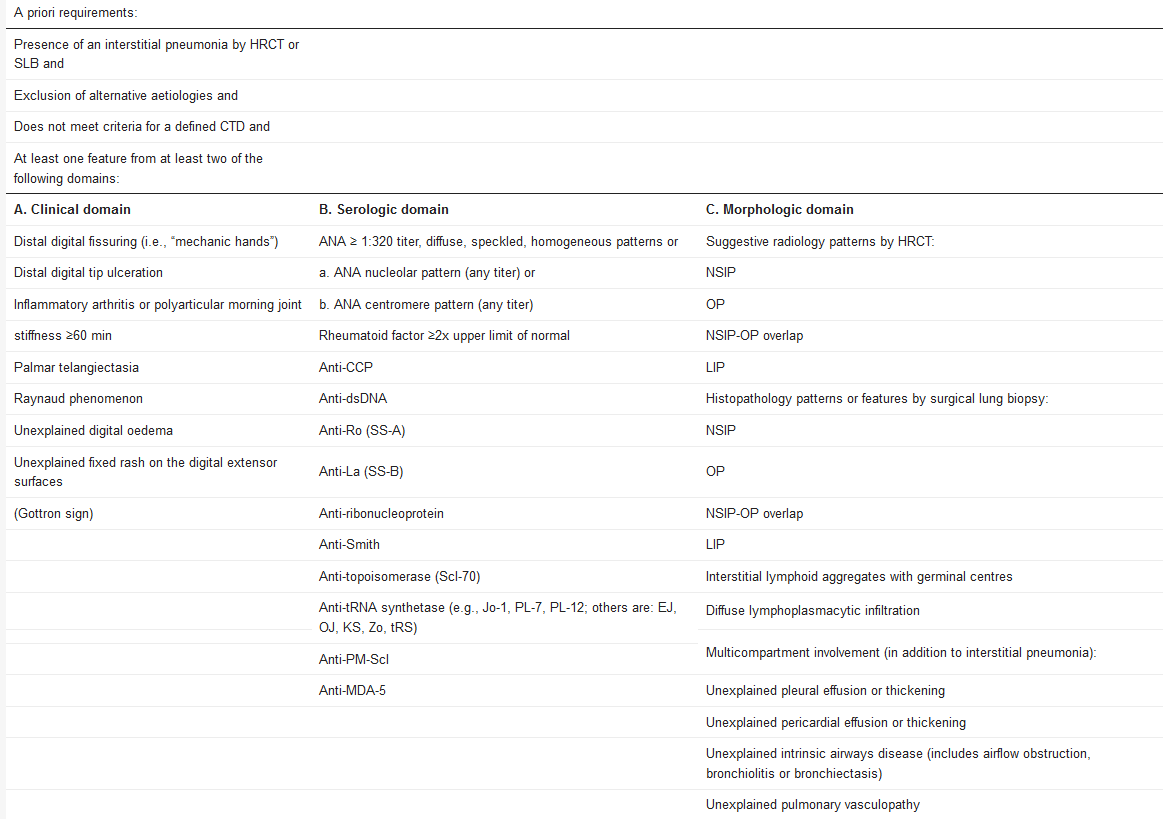
The included features were chosen for their higher specificity for CTDs. The term “connective tissue disease” was specifically avoided due to concerns that such labeling may give a false impression that these individuals have a defined CTD. In contrast, specific features suggesting vasculitis were deliberately excluded.
The Task Force included an international, multidisciplinary panel of CTD-ILD experts, including 13 pulmonologists, four rheumatologists, one thoracic radiologist, and one pulmonary pathologist. The proposed criteria reflected the panel’s expert opinion and Authors recognized that they should be tested and validated in future prospective studies, as a high-quality patient cohort for validation does not exist at the moment.